
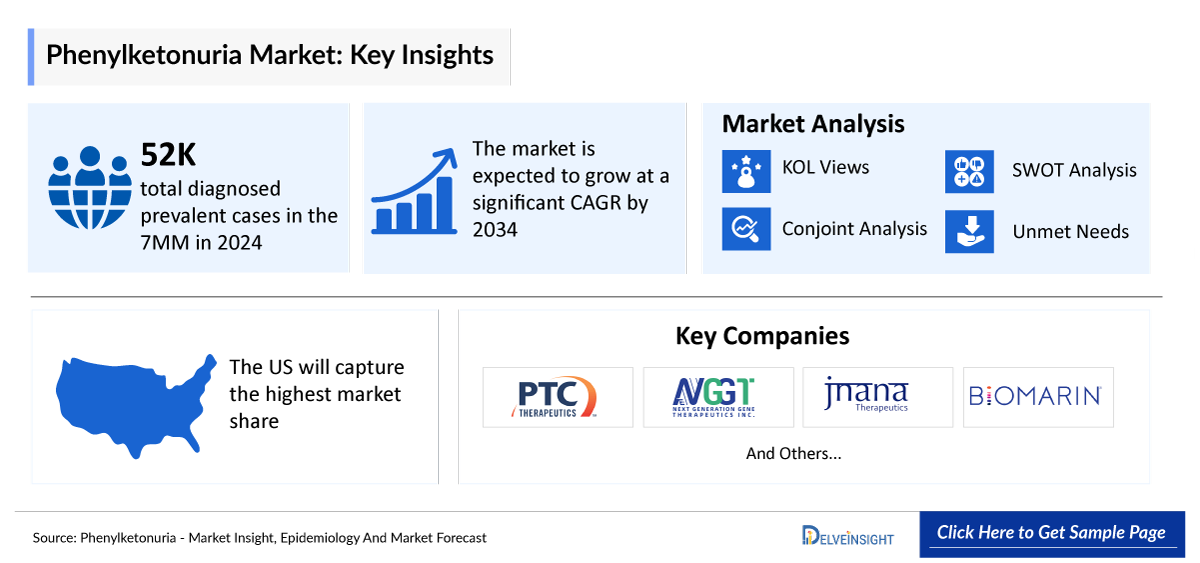
Phenylketonuria Market Summary
- The Phenylketonuria Market Size is estimated based on the study period 2022–2036 and is expected to grow during the forecast period (2026–2036).
- Early and accurate diagnosis of PKU remains a challenge, particularly in cases of early hospital discharge, which may lead to false negatives. Improved screening strategies that enhance sensitivity and specificity, including better evaluation of phenylalanine-to-tyrosine ratios and integration of genetic testing, are essential for timely and reliable identification of affected newborns.
- The leading Phenylketonuria companies developing therapies in the treatment market incude - PTC Therapeutics, Synlogic, and others.
Phenylketonuria Market and Epidemiology Analysis
- Among the 7MM, the United States had highest number of diagnosed prevalent cases of phenylketonuria (PKU) with 18,000 cases in 2025.
The age‐specific data revealed that the highest number of patients affected with PKU was found in the age group of <14 years. - Newborn blood testing identifies almost all cases of phenylketonuria. PKU is diagnosed by Phe and Tyr’s evaluation in neonatal dried blood spot (DBS) using tandem mass spectrometry. Bacterial inhibition assay (Guthrie test) is another simple, inexpensive, and reliable test used for many decades.
- Currently, the market landscape offers only two approved therapies, both of which are owned by BioMarin. The first is KUVAN (sapropterin), a synthetic form of tetrahydrobiopterin (BH4), which acts by increasing phenylalanine hydroxylase (PAH) activity. The second therapy is PALYNZIQ (pegvaliase), a phenylalanine ammonia-lyase (PAL) enzyme that temporarily restores the levels of PAH and reduces blood phenylalanine concentrations.
- The expected launch of potential therapies may increase market size in the coming years, assisted by an increase in the diagnosed prevalent population of PKU.
Request for unlocking the Sample Page of the "Phenylketonuria Market Insights"
Key Factors Driving the Phenylketonuria Market
The table given below further depicts the key segments provided in the report:
Scope of the Phenylketonuria Market | |
|
Study Period |
2022 to 2036 |
|
Forecast Period |
2026-2036 |
|
Geographies Covered |
|
|
Phenylketonuria Market |
|
|
Phenylketonuria Market Size | |
|
Phenylketonuria Companies |
PTC Therapeutics, Synlogic, and others |
|
Phenylketonuria Epidemiology Segmentation |
|
Key Factors Driving the Phenylketonuria Market
- Rising Diagnosed Patient Population: The diagnosed prevalent cases of PKU are increasing across the 7MM, with the US reporting 18,000 cases in 2025 and growth expected through 2036.
- Advancements in Pharmacological Therapies: Emerging therapies such as cofactor-based treatments and enzyme substitution therapies are improving metabolic control and expanding treatment options.
- Strong Pipeline and Innovation: Pipeline therapies including repinatrabit (JNT-517) and AG-181 are expected to transform the treatment landscape.
- Increased Newborn Screening: Improved neonatal screening programs have significantly enhanced early diagnosis and treatment outcomes.
- Shift Toward Targeted and Gene-based Therapies: Emerging strategies such as gene therapy and mRNA-based treatments aim to provide long-term disease control.
Phenylketonuria Disease Understanding
Phenylketonuria Overview
Phenylketonuria (PKU) is a rare genetic condition that causes an amino acid called phenylalanine to build up in the body; amino acids are the building blocks of protein. Phenylalanine is found in all proteins and some artificial sweeteners. PKU symptoms can range from mild to severe. The most severe form of this disorder is known as classic PKU. An infant with classic PKU may appear normal for the first few months. A less severe form of PKU is called variant PKU or non-PKU hyperphenylalaninemia. This occurs when the baby has too much phenylalanine in its body. Infants with this form of the disorder may have only mild symptoms.
Phenylketonuria Diagnosis
Phenylketonuria (PKU) diagnosis is achieved soon after birth by neonatal screening in most developed countries. In the countries where expanded newborn screening has been adopted, PKU is diagnosed by Phe and Tyr’s evaluation in neonatal dried blood spot (DBS) using tandem mass spectrometry. Bacterial inhibition assay (Guthrie test) is another simple, inexpensive, and reliable test used for many decades; however, it is a manual, semi-quantitative test and is being replaced by other methods in all screening laboratories. Some laboratories use a fluorimetric test that is quantitative, automated, and reliable.
Further details related to country-based variations are provided in the report…
Phenylketonuria Treatment
Sapropterin dihydrochloride, a synthetic form of tetrahydrobiopterin (BH4), has been introduced as a supplemental treatment to dietary Phe control for Phenylketonuria (PKU). BH4 is a naturally occurring compound cofactor for PAH and other enzymes. Several subsequent studies have found that BH4 supplementation effectively lowers blood serum Phe levels in some individuals with PKU.
KUVAN (sapropterin) is approved for PKU, which is a synthetic form of BH4, the cofactor for phenylalanine hydroxylase (PAH). PAH hydroxylates Phe through an oxidative reaction to form tyrosine. In patients with PKU, PAH activity is absent or deficient. Treatment with BH4 can activate residual PAH enzyme activity, improve the normal oxidative metabolism of Phe, and decrease Phe levels in some patients
Further details related to treatment and management are provided in the report…
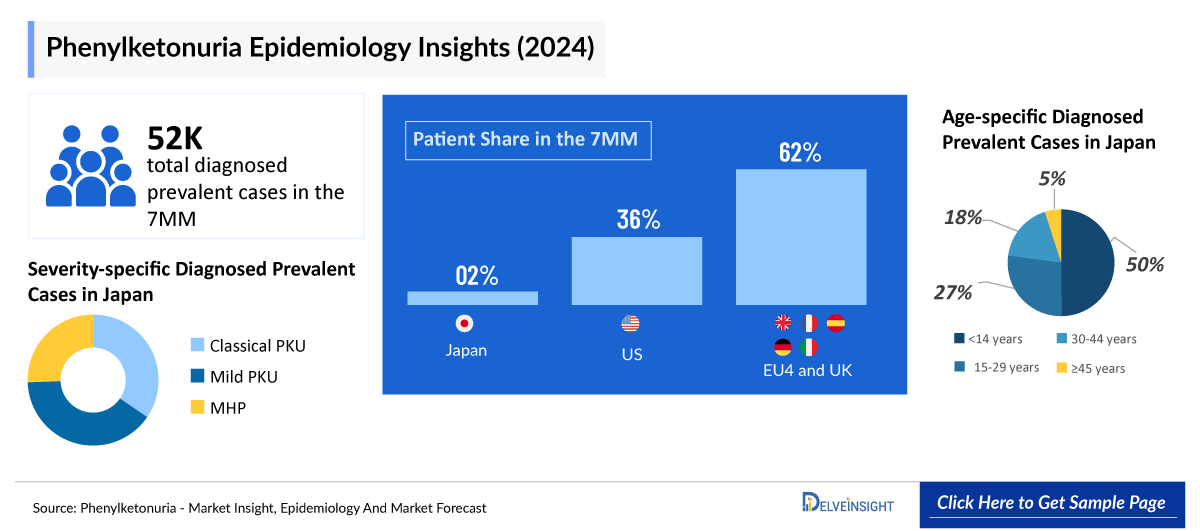
Phenylketonuria Epidemiology
The phenylketonuria epidemiology chapter in the report provides historical as well as forecasted epidemiology segmented by Total Diagnosed Prevalent Cases of PKU, Mutation Type-specific Cases of PKU, Age-specific Cases of PKU, and Severity-specific Cases of PKU in the United States, EU4 (Germany, France, Italy, Spain) and the United Kingdom, and Japan from 2022 to 2036.
Key Findings from Phenylketonuria Epidemiological Analyses and Forecast
- In 2025, the US recorded the highest number of diagnosed prevalent PKU cases among the 7MM, with 18,000 cases. This figure is projected to rise throughout the forecast period from 2026 to 2036.
- In 2025, the diagnosed prevalent cases of PKU across EU4 and the UK were approximately 32,000, with this number anticipated to grow by 2036.
- In Japan, the majority of PKU cases in 2025 were reported in the under-14 age group (530 cases), while the fewest cases (40) were observed in individuals aged 45 years and older.
- In 2025, the majority of severity-specific PKU cases in the US were attributed to classical PKU, accounting for 10,600 reported cases.
Phenylketonuria Epidemiology Segmentation
- Total Diagnosed Prevalent Cases of PKU
- Mutation Type-specific Cases of PKU
- Age-specific Cases of PKU
- Severity-specific Cases of PKU
Further details related to epidemiology will be provided in the report…
Phenylketonuria Recent Developments In The Treatment Market
-
In March 2026, Otsuka Pharmaceutical reported open-label extension data for repinatrabit (JNT-517), demonstrating a 67% reduction in phenylalanine levels
- In February 2026, BioMarin received FDA approval for an expanded indication of PALYNZIQ to include pediatric patients aged ≥12 years.
Phenylketonuria Drug Analysis
The section dedicated to drugs in the phenylketonuria report provides an in-depth evaluation of late-stage pipeline drugs (Phase III and Phase II) related to phenylketonuria pipeline drugs. The drug chapters section provides valuable information on various aspects related to clinical trials of phenylketonuria, such as the pharmacological mechanisms of the drugs involved, designations, approval status, patent information, and a comprehensive analysis of the pros and cons associated with each drug. Furthermore, it presents the most recent news updates and press releases on drugs targeting phenylketonuria.
Phenylketonuria Marketed Therapies
KUVAN (Sapropterin Hydrochloride): Asubio-Pharma/BioMarin-Pharmaceutical
KUVAN is indicated to reduce blood phenylalanine (Phe) levels in patients with hyperphenylalaninemia (HPA) due to tetrahydrobiopterin (BH4) responsive PKU and is to be used in conjunction with a Phe-restricted diet. In August 2021, NICE issued final draft guidance which recommends sapropterin (also called KUVAN) as an option for treating PKU in pregnant women until they give birth and for treating the condition in people until they turn 22.
PALYNZIQ (pegvaliase-pqpz/rAvPAL-PEG/BMN 165): BioMarin Pharmaceutical
PALYNZIQ (pegvaliase-pqpz) injection is the first FDA-approved enzyme substitution therapy for adults with PKU who have uncontrolled blood Phe levels above 600 µmol/L (10 mg/dL) on their current treatment. PALYNZIQ is a once-daily self-administered therapy that acts independently of the phenylalanine hydroxylase (PAH) enzyme, so it is an option for all eligible adult patients living with PKU. In October 2020, the US FDA approved the supplemental biologics license application to increase the maximum allowable dose of 60 mg with PALYNZIQ Injection for treating adults with PKU.
Phenylketonuria Emerging Therapies
Repinatrabit (JNT-517): Otsuka Pharmaceutical
Repinatrabit (JNT-517) is an investigational oral small-molecule therapy being developed by Jnana Therapeutics for the treatment of PKU. It is designed to selectively inhibit phenylalanine transport, thereby reducing systemic phenylalanine levels and improving metabolic control. The therapy represents a targeted approach aimed at addressing the underlying metabolic imbalance in PKU and is currently in Phase III.
In December 2025, Otsuka Pharmaceutical reported initiating a global Phase III trial of repinatrabit (JNT-517) for PKU; the therapy has received orphan drug and RPDD from the FDA. In August 2024, Otsuka Pharmaceutical reported entering into a definitive agreement to acquire Jnana Therapeutics for USD 800 million upfront, with up to USD 325 million in additional development and regulatory milestones. In September 2022, Jnana Therapeutics reported that JNT-517 received RPDD from the FDA for PKU, along with Orphan Drug Designation from the FDA and European Commission and eligibility recognition from the EMA.
AG-181: Agios Pharmaceuticals
AG-181 is an investigational small-molecule therapy being developed by Agios Pharmaceuticals for the treatment of PKU. It is designed to modulate metabolic pathways involved in phenylalanine regulation, aiming to reduce systemic phenylalanine levels and address the underlying metabolic imbalance in PKU. The therapy is currently in Phase I.
Note: Detailed assessment will be provided in the final report of Phenylketonuria…
Phenylketonuria Market Outlook
The goal of treatment for PKU is to keep plasma phenylalanine levels within 120−360 µmol/L (2−6 mg/dL). This is generally achieved through a carefully planned and monitored diet.
In 2007, the US FDA approved KUVAN (sapropterin hydrochloride) to treat PKU. KUVAN is an oral pharmaceutical formulation of BH4, the natural cofactor for the PAH enzyme, which stimulates the activity of the residual PAH enzyme to metabolize phenylalanine into tyrosine. KUVAN is to be used in conjunction with a phenylalanine-restricted diet. KUVAN is manufactured by BioMarin Pharmaceutical; however, it does not work for everyone with PKU. It is most effective in children with mild cases of PKU.
Researchers and other scientists are exploring additional treatments for PKU. These treatments include large neutral amino acid supplementation, which may help prevent phenylalanine from entering the brain, and enzyme replacement therapy, which uses a substance similar to the enzyme that usually breaks down phenylalanine.
Studies have shown that the treatment of PKU is multifaceted. The pipeline of PKU possesses a few potential drugs as monotherapies. Overall, the PKU therapeutics market is expected to grow in the forecast period (2024–2034).
Phenylketonuria Market Insights
- Various key players are leading the treatment landscape of phenylketonuria, such as Asubio-Pharma/BioMarin-Pharmaceutical, PTC Therapeutics, Synlogic, and others. The details of the country-wise and therapy-wise market size have been provided below.
- In 2023, the United States accounted for the largest market size among the 7MM, making ~60% of the total market size of the 7MM.
- Synlogic’s SYNB1934 is expected to become a key player of the PKU market during the forecast period (2026–2036).
- Among the EU4 and the UK, Germany had the largest market size with ~USD 70 million in 2023, while Spain had the smallest market size of PKU.
Phenylketonuria Competitive Landscape
The competitive landscape for Phenylketonuria (PKU) treatment and management is shaped by a combination of long-established dietary therapies, specialized medical foods, and innovative pharmaceutical solutions aimed at improving metabolic control and quality of life for patients.
- Dominance of medical foods and dietary management: Since strict dietary restriction of phenylalanine remains the backbone of PKU management, companies producing medical foods, low-protein formulas, and specialized nutritional products maintain a strong market presence. These products are essential for lifelong management and are widely prescribed globally.
- Emergence of targeted pharmacotherapy: In recent years, pharmaceutical companies have developed enzyme substitution and cofactor therapies (such as pegylated phenylalanine ammonia lyase and sapropterin dihydrochloride) that help lower blood phenylalanine levels in certain patients. These targeted therapies provide alternatives to dietary measures and expand treatment choices.
- Pipeline innovation: Biotech firms and specialty pharma companies are actively advancing gene therapies, novel enzyme therapies, and modified probiotics through clinical development. These next-generation approaches aim to offer more durable metabolic control or curative outcomes, creating differentiation in the competitive landscape.
- Strategic partnerships and acquisitions: Larger healthcare companies often engage in licensing agreements, collaborations, or acquisitions of emerging PKU assets to strengthen their portfolios and accelerate product development. These strategic moves influence market positioning and access to new technologies.
- Patient-centric services and digital health: Competitive differentiation also comes from support programs, patient education tools, digital tracking solutions, and nutritional counseling services that improve long-term adherence and outcomes, particularly for pediatric and adult populations.
Key Phenylketonuria Companies
The Key Phenylketonuria companies actively involved in the Phenylketonuria treatment landscape include -
- PTC Therapeutics
- Synlogic, and others
Phenylketonuria Drugs Uptake
This section focuses on the rate of uptake of the potential Phenylketonuria drugs recently launched in the Phenylketonuria treatment market or expected to get launched in the market during the study period 2020-2034. The analysis covers Phenylketonuria market uptake by drugs; patient uptake by therapies; and sales of each drug.
Phenylketonuria Drugs Uptake helps in understanding the drugs with the most rapid uptake, reasons behind the maximal use of new drugs, and allow the comparison of the drugs on the basis of Phenylketonuria market share and size which again will be useful in investigating factors important in market uptake and in making financial and regulatory decisions.
Phenylketonuria Pipeline Activities
The Phenylketonuria treatment market report provides insights into Phenylketonuria Clinical Trials within Phase II, and Phase III stage. It also analyses Phenylketonuria key players involved in developing targeted therapeutics.
Phenylketonuria Clinical Trials Activities
The Phenylketonuria treatment market report covers the detailed information of collaborations, acquisition, and merger, licensing, patent details, and other information for Phenylketonuria emerging therapies.
Latest KOL Views on Phenylketonuria Market Report
To stay abreast of the latest trends in the market, we conduct primary research by seeking the opinions of Key Opinion Leaders (KOLs) and Subject Matter Experts (SMEs) who work in the relevant field. This helps us fill any gaps in data and validate our secondary research.
We have reached out to industry experts to gather insights on various aspects of phenylketonuria, including the evolving treatment landscape, patients’ reliance on conventional therapies, their acceptance of therapy switching, drug uptake, and challenges related to accessibility. The experts we contacted included medical/scientific writers, professors, and researchers from prestigious universities in the US, Europe, the UK, and Japan.
Our team of analysts at Delveinsight connected with more than 15 KOLs across the 7MM. We contacted institutions such as the Lurie Children’s Hospital of Chicago, Ramón y Cajal Health Research Institute, Universitario Virgen del Rocío, Jichi Medical School, etc., among others. By obtaining the opinions of these experts, we gained a better understanding of the current and emerging treatment patterns in the phenylketonuria market, which will assist our clients in analyzing the overall epidemiology and market scenario.
Phenylketonuria Report Qualitative Analysis
We perform Qualitative and Market Intelligence analysis using various approaches, such as SWOT analysis and Conjoint Analysis. In the SWOT analysis, strengths, weaknesses, opportunities, and threats in terms of disease diagnosis, patient awareness, patient burden, competitive landscape, cost-effectiveness, and geographical accessibility of therapies are provided. These pointers are based on the Analyst’s discretion and assessment of the patient burden, cost analysis, and existing and evolving treatment landscape.
Conjoint Analysis analyzes multiple approved and emerging therapies based on relevant attributes such as safety, efficacy, frequency of administration, designation, route of administration, and order of entry. Scoring is given based on these parameters to analyze the effectiveness of therapy.
In efficacy, the trial’s primary and secondary outcome measures are evaluated; for instance, in trials for phenylketonuria, one of the most important primary endpoints were the percentage mean Phe (D5-Phe) reduction in plasma and fasting levels of plasma Phe. Based on these, the overall efficacy is evaluated.
Further, the therapies’ safety is evaluated wherein the acceptability, tolerability, and adverse events are majorly observed, and it sets a clear understanding of the side effects posed by the drug in the trials. In addition, the scoring is also based on the route of administration, order of entry and designation, probability of success, and the addressable patient pool for each therapy. According to these parameters, the final weightage score and the ranking of the emerging therapies are decided.
Phenylketonuria Market Access and Reimbursement Scenario
Because newly authorized drugs are often expensive, some patients escape receiving proper treatment or use off-label, less expensive prescriptions. Reimbursement plays a critical role in how innovative treatments can enter the market. The cost of the medicine, compared to the benefit it provides to patients who are being treated, sometimes determines whether or not it will be reimbursed. Regulatory status, target population size, the setting of treatment, unmet needs, the number of incremental benefit claims, and prices can all affect market access and reimbursement possibilities.
The report further provides detailed insights on the country-wise accessibility and reimbursement scenarios, cost-effectiveness scenario of approved therapies, programs making accessibility easier and out-of-pocket costs more affordable, insights on patients insured under federal or state government prescription drug programs, etc.
Scope of the Phenylketonuria Market Report
- The Phenylketonuria treatment market report covers the descriptive overview of Phenylketonuria, explaining its causes, signs and symptoms, pathophysiology, diagnosis and currently available Phenylketonuria therapies
- Comprehensive insight has been provided into the Phenylketonuria epidemiology and treatment in the 7MM
- Additionally, an all-inclusive account of both the current and emerging therapies for Phenylketonuria is provided, along with the assessment of new therapies, which will have an impact on the current treatment landscape
- A detailed review of the Phenylketonuria treatment market; historical and forecasted is included in the report, covering drug outreach in the 7MM
- The Phenylketonuria treatment market report provides an edge while developing business strategies, by understanding trends shaping and driving the global Phenylketonuria market
Phenylketonuria Market Report Summary
- The report offers extensive knowledge regarding the epidemiology segments and predictions, presenting a deep understanding of the potential future growth in diagnosis rates, disease progression, and treatment guidelines. It provides comprehensive insights into these aspects, enabling a thorough assessment of the subject matter.
- Additionally, an all-inclusive account of the current management techniques and emerging therapies and the elaborative profiles of late-stage (Phase III, Phase II, and Phase I) and prominent therapies that would impact the current treatment landscape and result in an overall market shift has been provided in the report.
- The report also encompasses a comprehensive analysis of the phenylketonuria market, providing an in-depth examination of its historical and projected market size (2020–2034). It also includes the market share of therapies, detailed assumptions, and the underlying rationale for our methodology. The report also includes drug outreach coverage in the 7MM region.
- The report includes qualitative insights that provide an edge while developing business strategies, by understanding trends, through SWOT analysis and expert insights/KOL views, including experts from various hospitals and prominent universities, patient journey, and treatment preferences that help shape and drive the 7MM phenylketonuria market.
Phenylketonuria Market Report Insights
- Phenylketonuria Patient Population
- Phenylketonuria Therapeutic Approaches
- Phenylketonuria Market Size and Trends
- Existing Market Opportunity
Phenylketonuria Market Report Key Strengths
- Eleven-year Forecast
- The 7MM Coverage
- Phenylketonuria Epidemiology Segmentation
- Patient-based Forecasting
- Key Cross Competition
Phenylketonuria Market Report Assessment
- Current Phenylketonuria Treatment Practices
- Reimbursements
- Phenylketonuria Market Attractiveness
- Qualitative Analysis (SWOT, Conjoint Analysis)
- Phenylketonuria Market Drivers
- Phenylketonuria Market Barriers
Key Questions Answered In The Phenylketonuria Market Report:
- Would there be any changes observed in the current treatment approach?
- Will there be any improvements in phenylketonuria management recommendations?
- Would research and development advances pave the way for future tests and therapies for phenylketonuria?
- Would the diagnostic testing space experience a significant impact and lead to a positive shift in the treatment landscape of phenylketonuria?
- What kind of uptake will the new therapies witness in coming years in phenylketonuria patients?

.png&w=256&q=75)


