Denali’s AVLAYAH Win for Hunter Syndrome Marks Turning Point for Rare Disease Innovation
Mar 30, 2026
Summary
- AVLAYAH (tividenofusp alfa‑eknm) has received FDA accelerated approval as the first Hunter syndrome therapy specifically designed to treat neurologic manifestations by crossing the blood–brain barrier.
- The drug uses Denali’s TransportVehicle platform to fuse the IDS enzyme with a transferrin‑receptor–targeting component, enabling enzyme delivery to both brain and peripheral tissues with a single weekly IV infusion.
- Approval is based on Phase 1/2 data showing deep reductions (around 90%) in cerebrospinal fluid heparan sulfate and normalization of this key biomarker in most treated children, a change judged reasonably likely to predict neurologic benefit.
- Continued approval depends on positive results from the ongoing Phase 2/3 COMPASS trial, which compares AVLAYAH against idursulfase and is designed to confirm meaningful cognitive, behavioral, and functional benefits in children with Hunter syndrome.
AVLAYAH’s U.S. approval marks a watershed moment for Hunter syndrome, delivering the first therapy specifically engineered to reach the brain and target the disease’s devastating neurologic manifestations in children. It also breaks a recent streak of rare-disease rejections at the FDA, sending an important signal about how regulators may handle innovative treatments built on strong biomarker science in ultra‑rare conditions.
Hunter syndrome (mucopolysaccharidosis type II, MPS II) is a rare X‑linked lysosomal storage disorder that primarily affects boys and is thought to impact roughly 500 individuals in the U.S. and about 2,000 worldwide. Mutations in the iduronate 2‑sulfatase (IDS) gene lead to deficient IDS enzyme activity and accumulation of glycosaminoglycans (GAGs) such as heparan sulfate and dermatan sulfate, causing progressive multi‑organ damage, including to the brain. Children can develop cognitive and behavioral decline, hearing loss, motor impairment, joint stiffness, and organ dysfunction, often beginning in early childhood.
Downloads
Click Here To Get the Article in PDF

Recent Articles
- FDA Approves Boehringer’s JASCAYD as First New IPF Therapy in Over a Decade; Denali Therapeutics&...
- Mucopolysaccharidosis: How the Different Types Affect Patients and Treatment Approaches?
- Merck’s WINREVAIR Granted FDA Priority Review for Pulmonary Arterial Hypertension; KalVista’s EKT...
- Rising of Orphan Drug Development
- Precigen’s PAPZIMEOS Wins Full FDA Approval for Recurrent Respiratory Papillomatosis; Tonix’s Ton...
Until now, Takeda’s idursulfase (ELAPRASE) was the only FDA‑approved treatment, providing systemic enzyme replacement but failing to cross the blood–brain barrier (BBB), and therefore offering no direct benefit for neurologic symptoms. For families watching children slowly lose speech, mobility, and cognition, the therapeutic landscape has long felt stagnant and painfully inadequate.
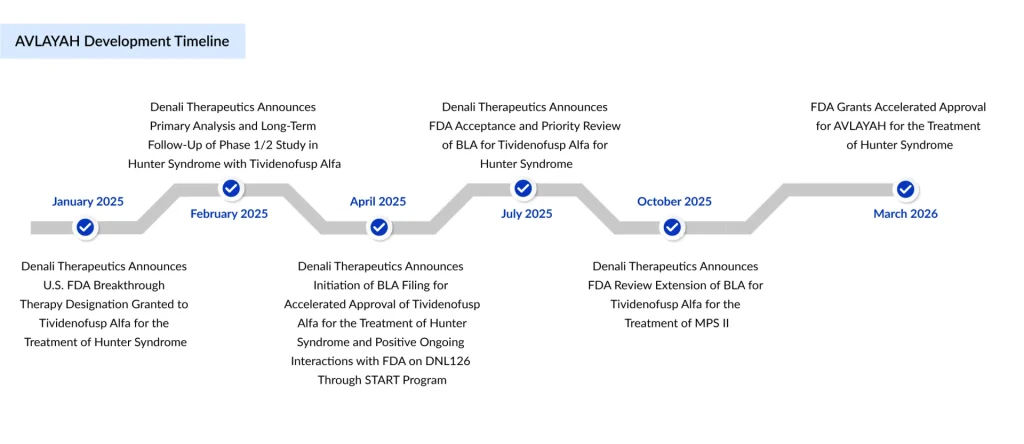
On March 25, 2026, the FDA granted accelerated approval to AVLAYAH (tividenofusp alfa‑eknm), an intravenous enzyme replacement therapy from Denali Therapeutics. AVLAYAH is indicated for the treatment of neurologic manifestations of Hunter syndrome when initiated in presymptomatic or symptomatic pediatric patients weighing at least 5 kg, before they reach advanced neurologic impairment.
This is the first new FDA‑approved treatment option for Hunter syndrome in nearly 20 years and the first product approved that is explicitly designed to address the disease’s neurologic complications. It is also the first FDA‑approved biotherapeutic that uses the transferrin receptor to cross the blood–brain barrier, inaugurating a new class of brain‑penetrant biologics.
The approval carries standard rare‑disease incentives and labels: AVLAYAH received Breakthrough Therapy, Fast Track, Orphan Drug, and Rare Pediatric Disease designations, and Denali was awarded a Rare Pediatric Disease Priority Review Voucher alongside approval.
AVLAYAH is built on Denali’s proprietary TransportVehicle (TV) platform, designed to shuttle large biologics into the brain by exploiting natural transport receptors on the BBB. The molecule fuses the IDS enzyme to an engineered Fc domain that binds the transferrin receptor (TfR) on endothelial cells lining the brain’s vasculature.
Once infused, AVLAYAH binds TfR, is ferried across the BBB via receptor‑mediated transcytosis, and then engages mannose‑6‑phosphate receptors on cells, delivering the IDS enzyme into lysosomes, where it can break down accumulated GAGs. Because TfR is widely expressed, the construct is expected to deliver IDS not only to the central nervous system but also to peripheral tissues, offering whole‑body and brain exposure in a single weekly infusion.
In contrast, legacy ERT, such as ELAPRASE, remains largely outside the CNS, improving somatic symptoms but leaving the neurologic trajectory largely unchanged. AVLAYAH’s brain‑penetrant design is therefore central to the excitement around its potential to alter the natural history of severe, neuronopathic MPS II.
The FDA’s decision rests on robust Phase 1/2 biomarker data rather than fully mature clinical outcomes, in keeping with the accelerated approval pathway. In an international, multi‑center open‑label trial, 47 children with Hunter syndrome (both ERT‑naïve and previously treated, ages 0.3 to 13 years) received weekly AVLAYAH infusions.
The primary objective was safety and tolerability, while secondary endpoints focused on central and peripheral pharmacodynamic effects, including heparan sulfate levels in cerebrospinal fluid (CSF) and urine, adaptive behavior measures, and liver volume. By week 24, AVLAYAH produced a 91% reduction in CSF heparan sulfate from baseline (95% CI: 89–92%), a magnitude of effect the FDA concluded was “reasonably likely to predict clinical benefit” on neurologic outcomes.
Notably, 93% of patients (41 of 44) had CSF heparan sulfate levels within the range observed in individuals without Hunter syndrome by week 24, suggesting normalization of this key disease biomarker in the vast majority of treated children. These data were compelling enough to earn publication in The New England Journal of Medicine on January 1, 2026, underscoring the scientific interest in this approach.
From a safety standpoint, the most common adverse events were infusion‑associated reactions, such as fever, chills, flushing, rash, and gastrointestinal symptoms, consistent with other ERTs. Three serious but manageable adverse events were reported in earlier datasets, and one patient discontinued treatment in part due to infusion reactions. Overall, the program met its predefined safety and tolerability expectations.
Despite its promise, AVLAYAH carries important risks that will shape how clinicians introduce the drug into practice. The U.S. prescribing information includes warnings for hypersensitivity reactions, including life‑threatening anaphylaxis, which have occurred both early and after multiple doses, with symptoms such as wheezing, hypotension, hives, vomiting, and swelling of the lips or tongue.
Infusion‑associated reactions can occur during or within 24 hours of dosing and may include chills, fever, flushing, rash, low blood pressure, tachycardia, respiratory symptoms, and gastrointestinal complaints, sometimes necessitating premedication, slowing or pausing the infusion, or stopping treatment in severe cases. Anemia has been observed and may require periodic monitoring of hemoglobin, while membranous nephropathy has been reported in at least one patient, prompting recommendations for ongoing renal function surveillance.
The label states that AVLAYAH is not recommended in combination with other enzyme replacement therapies, leaving physicians to consider switching rather than layering therapies for patients already on Elaprase. Overall, the safety profile appears clinically manageable in experienced hands but will require structured infusion protocols and careful monitoring, especially in children with underlying cardiac or pulmonary compromise.
Continued FDA approval is contingent on verification of clinical benefit in the ongoing Phase 2/3 COMPASS trial, which is already more than 95% enrolled according to regulators. This randomized study compares AVLAYAH head‑to‑head with idursulfase in children and young adults with Hunter syndrome, using a 2:1 randomization schema in favor of AVLAYAH.
COMPASS is designed to capture cognitive, adaptive behavior, and functional outcomes across the MPS II spectrum, aiming to translate biomarker normalization into measurable preservation of development, behavior, and quality of life. Denali intends to use COMPASS as the backbone for global regulatory submissions, and the European Medicines Agency has already granted Priority Medicines (PRIME) designation to tividenofusp alfa, underscoring its potential impact in an area of high unmet need.
Until full clinical data mature, the field will watch closely to see whether deep reductions in CSF heparan sulfate truly correlate with slowed neurocognitive decline and improved daily functioning in real‑world children.
AVLAYAH’s approval lands against the backdrop of a turbulent period for rare‑disease drug developers, marked by a series of high‑profile FDA rejections and shifting expectations around evidence standards. Earlier in 2026, the agency issued a complete response letter to REGENXBIO’s Hunter syndrome gene therapy, citing concerns about the use of natural‑history controls, eligibility criteria, and biomarker validation, despite prior dialogue that suggested the design might be acceptable.
Those decisions fueled anxiety that regulators were becoming less flexible on rare‑disease programs, particularly in ultra‑rare, pediatric neurodegenerative settings where large randomized trials can be difficult or impossible. In that context, AVLAYAH’s accelerated approval based on a rigorously characterized surrogate endpoint is being viewed as a “welcome positive” and a sign that the FDA can still exercise appropriate flexibility when the totality of biomarker, mechanistic, and clinical data is compelling.
In conclusion, AVLAYAH’s approval is not the end of the story; it is a beginning. The COMPASS trial must still demonstrate that biomarker gains translate into meaningful clinical benefits, regulators outside the U.S. must weigh the data under their own frameworks, and payers will have to decide how to reimburse a high‑cost, first‑in‑class therapy for an ultra‑rare disease.
But from a scientific and regulatory standpoint, the precedent is powerful: an engineered enzyme replacement therapy that crosses the blood–brain barrier, normalizes a central biomarker in the vast majority of treated children, and wins accelerated approval in an otherwise hostile rare‑disease climate. For Hunter syndrome and for the broader field of neuro‑rare disorders, AVLAYAH’s approval is more than a label; it is a signal that with the right science and advocacy, the bar for meaningful progress, brain included, can be cleared.

Downloads
Article in PDF
Recent Articles
- Precigen’s PAPZIMEOS Wins Full FDA Approval for Recurrent Respiratory Papillomatosis; Tonix’s Ton...
- Unveiling Lysosomal Storage Disorders: Exploring Rare Diseases Impacting Millions Worldwide
- FDA Approves Boehringer’s JASCAYD as First New IPF Therapy in Over a Decade; Denali Therapeutics&...
- Mucopolysaccharidosis: How the Different Types Affect Patients and Treatment Approaches?
- Rising of Orphan Drug Development



