Unveiling Lysosomal Storage Disorders: Exploring Rare Diseases Impacting Millions Worldwide
May 21, 2025
Table of Contents
Lysosomal Storage Disorders, or LSDs, are rare genetic conditions that may not make headlines, but they affect thousands worldwide, often with life-changing consequences. LSDs happen when the body’s lysosomes, tiny cellular recycling centers, fail to break down waste materials due to missing or faulty enzymes. This leads to a buildup of harmful substances, causing damage to organs like the brain, heart, and bones. Globally, LSDs impact about 1 in 5K to 1 in 10K people, with over 70 distinct disorders identified, totaling around 700K patients worldwide.
Living with an LSD can be tough. Symptoms vary widely, from developmental delays and seizures to bone deformities and organ failure, often appearing in childhood but sometimes later in life. While individually rare, their collective burden is significant, with annual treatment costs reaching billions globally. Advances in enzyme replacement therapies, gene therapies, and early diagnosis are offering hope, but challenges like high costs and limited access persist, especially in low-income regions.
Downloads
Click Here To Get the Article in PDF

Recent Articles
- Predicting risk of Acute Myeloid Leukemia (AML) in healthy individuals
- AZ announces positive results for durvalumab and tremelimumab; FDA nods to extend LILETTA’s use t...
- Ethicon’s ETHIZIA Hemostatic Sealing Patch; FDA Approves Medtronic’s Minimally Invasive Device to...
- Gaucher Disease: “Rare Disorder with High Unmet Needs”
- AI Stethoscope: Transforming the Stethoscope Market and Cardiac Diagnostics with Unmatched Accuracy
Types of Lysosomal Storage Disorders
Lysosomal Storage Disorders are a group of over 70 rare, inherited metabolic conditions caused by enzyme deficiencies that prevent the proper breakdown of waste materials inside cells. This dysfunction leads to the toxic buildup of substances, resulting in progressive damage to multiple organs and systems. While the underlying cellular defect is similar, LSDs vary widely in their clinical presentation, severity, and treatment needs.

Some of the most recognized types include Gaucher Disease, Fabry Disease, Niemann-Pick Disease Types A/B and C, Krabbe Disease, Metachromatic Leukodystrophy (MLD), GM1 Gangliosidosis, Tay-Sachs Disease, Sandhoff Disease, Mucopolysaccharidoses (MPS) Types I, II (Hunter Syndrome), and III (Sanfilippo Syndrome), Pompe Disease, Cystinosis, Danon Disease, and Wolman Disease.
Let’s take a closer look at these disorders, how they differ, the burden they impose, and the market landscape that continues to evolve. With innovations in enzyme replacement, gene therapy, and early diagnostics, the future of LSD treatment is steadily advancing, offering renewed hope for patients and caregivers around the world.
Gaucher’s Disease
Gaucher’s Disease is a rare, inherited lysosomal storage disorder caused by a deficiency of the enzyme glucocerebrosidase, due to mutations in the GBA1 gene. This deficiency causes glucocerebroside to accumulate in organs like the spleen, liver, and bone marrow, leading to symptoms such as anemia, bone pain, fatigue, and organ enlargement. Gaucher’s disease presents in three types, with Type 1 being the most common and non-neuronopathic, while Types 2 and 3 involve severe neurological complications. According to the National Institutes of Health, Gaucher’s disease affects about 1 in 40K to 1 in 100K people worldwide, equating to approximately 70K to 175K cases globally. The National Organization for Rare Disorders notes a higher Gaucher disease prevalence among Ashkenazi Jewish populations, where incidence can reach 1 in 450, driving targeted genetic screening efforts.
There is no cure for Gaucher’s disease, but enzyme replacement therapy and substrate reduction therapy are available to mitigate manifestations. Enzyme replacement therapies, administered via IV infusions, include CEREZYME by Sanofi Genzyme, approved by the FDA in 1994, VPRIV by Takeda, approved in 2010, and ELELYSO by Pfizer and PROTALIX, approved in 2012. These Gaucher’s disease drugs replenish the deficient enzyme, reducing organ enlargement and improving blood counts in Type 1 and non-neurological Type 3 cases. For patients unsuitable for ERT, oral substrate reduction therapies like ZAVESCA by Actelion, the first oral agent approved in 2003 for mild to moderate Type 1 Gaucher’s disease, and CERDELGA by Sanofi, approved in 2014, offer alternatives. These treatments, while effective for systemic Gaucher’s disease symptoms, do not address neurological complications, highlighting a significant unmet need in the therapeutic landscape.
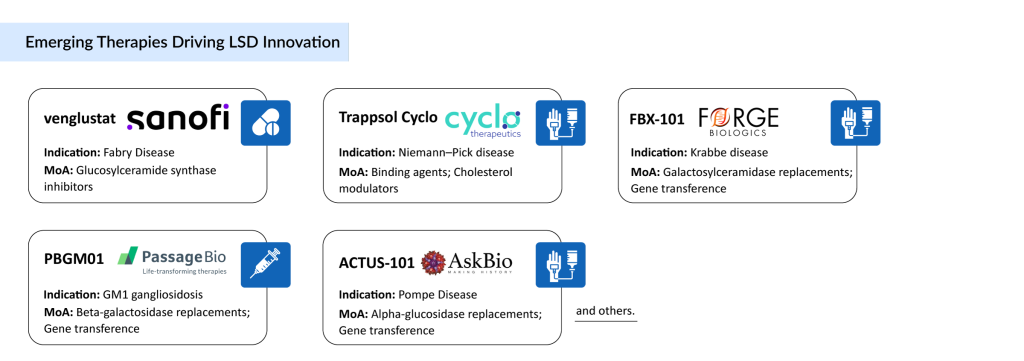
The pipeline for Gaucher’s disease includes several promising candidates, such as Venglustat and GZ385660 by Sanofi, FLT201 by Spur Therapeutics, LY3884961 by Prevail Therapeutics, and YH35995 by Yuhan Corporation. These emerging Gaucher’s disease therapies, spanning oral small molecules and gene therapies, aim to address both systemic and neuropathic complications, potentially revolutionizing the Gaucher disease treatment market. Growing awareness about Gaucher’s disease and its treatment options, increasing R&D expenditures, and a robust pipeline are fueling innovation, with gene therapies offering hope for tackling the root cause of the disease.
The Gaucher disease market is poised for growth across the 7MM, driven by increasing prevalence and improved diagnosis of Gaucher’s disease. Growing awareness through initiatives like the National Gaucher’s Foundation and rising R&D investments are key factors supporting the Gaucher disease treatment market. The high unmet need for therapies that combat neuropathic complications at reduced costs and with convenient administration schedules continues to drive research. As emerging Gaucher’s disease therapies progress through clinical development and diagnostic rates improve, the therapeutic landscape is expected to expand significantly, promising better outcomes for patients worldwide.
Explore a comprehensive analysis of the Gaucher Disease Market in our latest report.
Fabry Disease
Fabry Disease is a rare, X-linked lysosomal storage disorder caused by mutations in the GLA gene, leading to a deficiency of the enzyme alpha-galactosidase A. This deficiency causes globotriaosylceramide (Gb3) to build up in organs like the kidneys, heart, and nervous system, resulting in symptoms such as burning pain in the hands and feet, kidney dysfunction, heart complications, and skin lesions. Fabry disease affects both males and females, though symptoms are often milder in females due to X-chromosome mosaicism.
Based on DelveInsight’s assessment in 2024, the 7MM had approximately 18K diagnosed prevalent cases of Fabry disease. In the 7MM, the US accounted for the highest number of diagnosed prevalent cases, with nearly 9.2K cases in 2024. In the US, the 10 to 19 years age group had the highest number of cases, while the under 10 years and 50 years and older age groups accounted for the least number of cases. The global burden of Fabry disease has increased over the years, and with the introduction of newborn screening, the estimation of actual prevalence has improved significantly, aiding early diagnosis and treatment initiation.
Treatment of Fabry disease consists of enzyme replacement therapies, oral chaperone therapy, and adjunctive treatments like ACE inhibitors, antiplatelet drugs, and analgesics. Currently, three therapies are approved for treating patients with Fabry disease in the US, including ELFABRIO by Chiesi and Amicus, GALAFOLD by Amicus Therapeutics, and FABRAZYME by Sanofi Genzyme. In Europe, four therapies are approved, including ELFABRIO, REPLAGAL by Takeda, FABRAZYME, and GALAFOLD, while Japan has REPLAGAL and GALAFOLD. GALAFOLD, an oral capsule, is gaining traction, with many patients switching from ERTs due to its convenience and clinical benefits, as noted by the National Institute for Health and Care Excellence for its cost-effectiveness. These Fabry disease drugs can delay complications, but limitations, including infusion reactions and incomplete organ protection, highlight the need for innovative treatments.
Several companies are working to develop new Fabry disease therapies with novel mechanisms to address unmet needs in the therapeutic landscape. Emerging therapies include venglustat by Sanofi Genzyme, lucerastat by Idorsia Pharmaceuticals, ST-920 by Sangamo Therapeutics, 4D-310 by 4D Molecular Therapeutics, and AMT-191 by UniQure. These candidates, particularly gene therapies like ST-920, which has shown promise in raising enzyme levels and clearing Gb3 in animal models, are expected to enter the Fabry disease market during the forecast period. The dearth of late-stage pipeline candidates underscores the challenge of developing transformative treatments, but these therapies hold potential to reshape the Fabry disease treatment market.
The total market size in the 7MM for Fabry disease was nearly USD 1.7 billion in 2024, with the US holding the largest share. DelveInsight expects the Fabry disease market to grow positively by 2034, driven by increasing prevalence, improved diagnosis through newborn screening, and the launch of upcoming therapies. The shift toward GALAFOLD and the anticipated entry of gene therapies like ST-920, projected to generate the highest revenue among new therapies, will fuel growth. Despite challenges like high treatment costs, the Fabry disease treatment market is poised for expansion, offering hope for better management of symptoms and improved patient outcomes globally.
Gain deep insights into the Fabry Disease Market through our expert-driven research.
Niemann-Pick Disease
Niemann-Pick Disease is a rare group of inherited lysosomal storage disorders that disrupt the body’s ability to process lipids, leading to harmful accumulations in organs like the liver, spleen, and brain. This causes a range of symptoms, from developmental delays and seizures to liver failure and neurological decline, often striking in childhood but sometimes later in life. Niemann-Pick Disease is caused by genetic mutations that impair lipid metabolism, making it a devastating condition for affected families worldwide. According to the National Institutes of Health, Niemann-Pick Disease affects approximately 1 in 120K to 1 in 250K people globally, equating to about 30K to 65K cases worldwide. With growing awareness and genetic testing, the need for effective Niemann-Pick Disease treatments is more urgent than ever.
Niemann-Pick Disease Types A and B
Niemann-Pick Disease Types A and B are caused by mutations in the SMPD1 gene, which leads to a deficiency in the enzyme acid sphingomyelinase. Type A is more severe and primarily affects infants, often resulting in death by early childhood. It is especially prevalent among Ashkenazi Jews. Symptoms include rapid neurodegeneration, hepatosplenomegaly, and failure to thrive.
Type B, by contrast, is less severe and does not typically involve the central nervous system. It can be diagnosed later in childhood or even adulthood, with patients potentially living into adulthood. Type B presents with enlarged liver and spleen, lung disease, and low blood platelet counts, but usually without severe neurological decline.
Niemann-Pick Disease Type C
Niemann-Pick Disease Type C (NPC) is caused by mutations in the NPC1 or NPC2 genes and involves a defect in intracellular cholesterol trafficking. It is clinically distinct from Types A and B and can vary significantly in its onset and progression. Symptoms may include ataxia, cognitive decline, psychiatric symptoms, seizures, and organ dysfunction, and onset can occur at any age, from infancy to adulthood. Of the gene-specific subtypes, NPC1 accounts for the majority of cases.
According to the analysis conducted by DelveInsight, in 2023, the total diagnosed prevalent cases of Niemann-Pick Disease Type C in the 7MM were nearly 789, with these cases projected to increase during the forecast period from 2025 to 2034. In Japan, approximately 37 diagnosed prevalent cases of juvenile NPC (ages 6 to under 15 years) were reported in 2023. Across the 7MM, diagnosed prevalent cases varied by gender, with higher prevalence observed in females (65%) compared to males (45%).
Treatment of Niemann-Pick Disease Type C requires a multidisciplinary approach, as no current therapy has proven capable of halting disease progression. Currently, two therapies are approved in the US:
- AQNEURSA (levacetylleucine) by IntraBio: For neurological symptoms in patients weighing at least 15 kg.
- MIPLYFFA (arimoclomol) by Zevra Therapeutics: Approved for neurological symptoms in both children and adults.
To dive deeper into MIPLYFFA and AQNEURSA, explore our dedicated blog that breaks down everything you need to know about these two promising treatments. Click here to read the full blog.
Miglustat is approved in Europe (since 2009) and Japan (since 2012) for NPC, though it is used off-label in the US after the FDA declined approval in 2010. Miglustat is also authorized for Gaucher disease.
Symptomatic therapies, such as speech therapy and gastrostomy tubes, play a critical role in managing daily functions and improving quality of life.
While no late-stage candidates are currently in the Niemann-Pick pipeline, multiple companies across the 7MM are actively exploring new therapeutic options. These emerging treatments are expected to enter the market during the forecast period, potentially offering novel mechanisms to combat both neurological and visceral symptoms. The lack of late-stage therapies underscores the developmental challenges for this rare condition but highlights a growing commitment to improve the Niemann-Pick Disease treatment landscape.
In 2023, the total market size in the 7MM for NPC was approximately USD 34 million, with the US accounting for nearly USD 9 million. DelveInsight forecasts that the market will grow at a significant CAGR through 2034, driven by rising diagnosed prevalence and the launch of emerging therapies. Continued advancements in genetic screening and increased symptom awareness are key to unlocking future market potential. Despite the high costs and limited therapeutic arsenal, the market is gradually evolving toward better patient outcomes and quality of life enhancements worldwide.
Stay updated with the evolving landscape of the Niemann-Pick Disease C Market in our in-depth report.
Krabbe disease
Krabbe Disease is a rare, inherited lysosomal storage disorder caused by mutations in the GALC gene, leading to a deficiency of the enzyme galactocerebrosidase. This deficiency causes toxic buildup of psychosine, damaging the nervous system and resulting in severe symptoms like muscle stiffness, developmental delays, seizures, vision loss, and hearing impairment. Krabbe Disease primarily affects infants, with most cases appearing before six months, though late-onset forms can emerge in childhood or adulthood. The relentless progression of Krabbe Disease symptoms devastates families, making early diagnosis and treatment critical.
According to the National Organization for Rare Disorders in 2024, an estimated 1 in 100K newborn babies have Krabbe Disease, with the severe infantile-onset form occurring in 85 to 90% of patients, while late-onset cases account for 10 to 15%, potentially higher based on state newborn screening data. Metabolic Support UK notes a similar prevalence of 1 in 100K in Northern European populations. These figures highlight the global burden of Krabbe Disease, with screening programs improving case detection, particularly for late-onset forms, which may enable earlier intervention to manage the devastating Krabbe Disease symptoms.
No therapies are currently approved by the FDA for Krabbe Disease, but hematopoietic stem cell transplantation remains the gold standard treatment. For early-onset Krabbe Disease, HSCT is recommended in asymptomatic infants under one month old, while late-onset cases require symptom monitoring over 3 to 6 months to assess progression before transplantation. HSCT can slow disease progression by providing functional GALC enzyme through donor cells, but its efficacy depends on early intervention and patient suitability, determined through rigorous neurological and physical examinations. Symptomatic treatments, such as physical therapy for muscle stiffness and anticonvulsants for seizures, are critical components of the Krabbe Disease therapeutic landscape, though they do not address the underlying cause.
Several companies are developing novel Krabbe Disease therapies to address unmet needs, with promising candidates in early to mid-stage clinical development. Key pipeline therapies include FBX-101 by Forge Biologics and PLX-300 by Polaryx Therapeutics, both exploring gene therapy to restore GALC levels. Other research efforts focus on substrate reduction therapies, like acid ceramidase inhibitors, and viral gene therapies, such as AAV-based approaches combined with bone marrow transplantation, which have shown significant improvements in preclinical models like Twitcher mice. Companies like Pfizer, Sanofi-Aventis SA, Shire, GlaxoSmithKline, Johnson & Johnson, Abbott Laboratories, and others are also involved in the broader Krabbe Disease pipeline, investigating combinations of gene therapy, enzyme replacement, and anti-inflammatory treatments. These emerging Krabbe Disease therapies aim to transform the treatment landscape by targeting both central and peripheral nervous system pathology.
The Krabbe Disease market is projected to grow steadily from 2020 to 2034, driven by rising awareness, improved diagnostic capabilities, and expanding research in gene and enzyme replacement therapies. Promising candidates like FBX-101 and PLX-300, along with advancements in combination therapies, are enhancing treatment prospects. Increased newborn screening, coupled with strong advocacy and investment, is further accelerating therapeutic development. Despite its low incidence, the market is set for meaningful growth, with innovative approaches aiming to improve outcomes for Krabbe Disease patients globally by 2034.
Discover breakthrough trends and future outlooks in the Krabbe Disease Market with our latest publication.
Metachromatic Leukodystrophy (MLD)
Metachromatic Leukodystrophy is a rare inherited lysosomal storage disorder caused by a deficiency of the enzyme arylsulfatase A (ARSA). This deficiency leads to the accumulation of sulfatides in the central and peripheral nervous systems, causing progressive demyelination, or the loss of the protective myelin sheath around nerve cells. MLD mainly affects children and is categorized into three subtypes based on the age of onset: late-infantile, juvenile, and adult. Symptoms may include motor skill regression, cognitive decline, and spasticity in children, while adults often experience psychiatric symptoms and dementia.
Traditionally, treatment options for MLD were limited to supportive care aimed at symptom management. Hematopoietic stem cell transplantation (HSCT) has shown some benefit, particularly when administered before symptoms appear, but it comes with high risks and unpredictable outcomes. A major turning point in treatment came with the emergence of gene therapy.
The market for MLD therapies, though small due to the ultra-rare nature of the disease, has seen increased attention. This growth is driven by the high unmet need and the promise of long-term benefits from precision medicine approaches like gene therapy. Payers and health systems face challenges with the cost, but strong advocacy exists for reimbursement due to the life-threatening nature of the condition. Biotech companies are showing growing interest, particularly in North America and Europe, with ongoing research into next-generation gene therapies and potential enzyme replacement therapies.
Emerging treatments include SHP611 and HGT-1110 from Shire, alongside promising programs from companies such as Takeda, Denali Therapeutics, Orchard Therapeutics, Homology Medicines, Passage Bio, ArmaGen Technologies, and others. These players are actively advancing the MLD therapeutic pipeline to transform outcomes for affected patients.
Looking ahead, the MLD treatment landscape is likely to improve with the expansion of newborn screening programs that include genetic testing. This will enable earlier diagnosis and make early intervention more viable. Regulatory incentives for orphan drugs, along with advancements in gene therapy development and delivery methods, may help reduce costs and broaden access. The commercialization of MLD therapies is also setting the stage for innovation across the broader rare disease sector, positioning it as a model for future therapeutic breakthroughs.
Delve into expert perspectives and pipeline evaluations in the Metachromatic Leukodystrophy (MLD) Market.
GM1 Gangliosidosis
GM1 Gangliosidosis is a rare, inherited, autosomal recessive lysosomal storage disorder caused by mutations in the GLB1 gene, leading to a deficiency of the enzyme beta-galactosidase. This deficiency causes ganglioside GM1 and other substances to accumulate in the brain, nervous system, and other organs, triggering severe symptoms like developmental regression, seizures, muscle weakness, vision and hearing loss, and organ enlargement. GM1 Gangliosidosis primarily affects infants, with the severe infantile form progressing rapidly, though juvenile and adult-onset forms exist with milder symptoms. The devastating impact of GM1 Gangliosidosis symptoms, often leading to early mortality in infantile cases, underscores the urgent need for effective treatments.
As per secondary research, GM1 Gangliosidosis is an autosomal recessive lysosomal storage disorder affecting 1 in every 100K to 200K live births within the general population. The National Organization for Rare Disorders confirms this prevalence, noting that the infantile form accounts for the majority of cases, while juvenile and adult-onset forms are rarer but increasingly detected through improved genetic screening. The global burden of GM1 Gangliosidosis is significant, with enhanced diagnostic capabilities and newborn screening programs improving case identification, particularly for later-onset forms, which may enable earlier intervention to manage GM1 Gangliosidosis symptoms.
There is currently no cure for GM1 Gangliosidosis, and no FDA-approved drugs directly target the underlying cause of the disease. GM1 Gangliosidosis treatment is primarily supportive, focusing on managing symptoms to enhance patients’ quality of life. Anticonvulsant medications like gabapentin are used to control seizures, while gastrostomy tubes ensure proper nutrition and hydration for those with swallowing difficulties. Physical and occupational therapy help maintain muscle strength and mobility, and respiratory support may be needed in advanced cases. Some patients benefit from a ketogenic diet to reduce seizure frequency. These symptomatic treatments, while critical for the GM1 Gangliosidosis therapeutic landscape, do not alter the disease’s progressive course, highlighting the need for disease-modifying therapies.
The development pipeline for GM1 Gangliosidosis therapies remains limited but promising, with gene therapy emerging as a key focus. Among the leading candidates is Passage Bio’s PBGM01, a gene therapy designed to deliver a functional GLB1 gene, currently in Phase I/II clinical trials. In August 2024, Passage Bio out-licensed PBGM01 to GEMMA Biotherapeutics, granting exclusive worldwide rights for its development and commercialization—a strategic move expected to accelerate innovation in the GM1 Gangliosidosis treatment market.
While the pipeline is still in early stages and lacks approved therapies, DelveInsight projects steady growth in the GM1 Gangliosidosis market across the 7MM from 2025 to 2034, fueled by improved diagnostics, rising disease awareness, and early case detection. Despite ongoing challenges such as high development costs and limited treatment options, the focus on emerging GM1 Gangliosidosis therapies, particularly gene therapies, signals a transformative shift toward addressing the root cause of the disease and offers hope for a more effective and sustainable treatment future.
Analyze emerging therapies and market dynamics in the GM1 Gangliosidosis Market in our comprehensive report.
Tay-Sachs Disease
Tay-Sachs Disease is a rare, inherited neurodegenerative lysosomal storage disorder caused by mutations in the HEXA gene, resulting in a deficiency of the enzyme hexosaminidase A. This deficiency leads to the accumulation of GM2 ganglioside in nerve cells of the brain and spinal cord, causing progressive damage and symptoms like seizures, muscle weakness, vision and hearing loss, and developmental regression. Tay-Sachs disease primarily affects infants, with the severe infantile form being the most common, though rarer juvenile and adult-onset forms exist with milder symptoms. The devastating impact of Tay-Sachs disease symptoms, often leading to early mortality in infantile cases, makes early diagnosis and intervention critical.
As per the National Organization for Rare Disorders, Tay-Sachs Disease has an estimated global prevalence of 1 in 320K live births, with significantly higher rates in specific communities, such as Ashkenazi Jewish, French Canadian, and Cajun populations, where carrier screening has improved detection. Newborn screening and genetic testing are increasing case identification, particularly for late-onset forms, which may enable earlier management of Tay-Sachs disease symptoms. The global burden of this disorder remains significant, with advocacy efforts and rare disease initiatives highlighting the need for effective treatments.
There is currently no cure for Tay-Sachs disease, and no FDA-approved drugs directly target the underlying cause of the disease. Treatment focuses on supportive care to manage symptoms and improve quality of life. Anticonvulsant medications, such as gabapentin, help control seizures, while physical therapy maintains muscle strength and mobility. Nutritional support, including gastrostomy tubes, ensures adequate hydration and nutrition for patients with swallowing difficulties. Respiratory support may be required in advanced cases to address breathing challenges. These symptomatic treatments, essential to the Tay-Sachs market, do not alter the disease’s progressive course, underscoring the urgent need for disease-modifying Tay-Sachs disease drugs.
The pipeline for Tay-Sachs disease includes promising research into gene therapy, enzyme replacement therapy, and substrate reduction therapy. Gene therapy, aimed at delivering a functional HEXA gene to correct the enzyme deficiency, is in early-stage clinical trials, showing potential in preclinical models to slow GM2 ganglioside accumulation. Enzyme replacement therapy, which seeks to supplement the missing hexosaminidase A enzyme, is under investigation but not yet available. Substrate reduction therapy, designed to reduce GM2 ganglioside production, is also being explored, though it remains in early development. Companies like Genzyme, Sanofi, Azafaros A.G., Idorsia Pharmaceuticals, Taysha Gene Therapies, GlycoNet, IntraBio, Actelion, Natera, are driving these Tay-Sachs disease therapies, but the pipeline is limited, reflecting the challenges of developing treatments for such a rare condition. These efforts hold promise for transforming the Tay-Sachs disease treatment market.
The Tay-Sachs disease market is relatively small due to the rarity of the condition, but it is growing with advances in genetic and enzyme-based therapies. DelveInsight projects market growth in the 7MM, driven by increased awareness, better diagnostics, and potential new treatments. Rising focus on orphan drugs, supported by funding and partnerships, is also boosting the market. However, high development costs and limited access to specialized care remain challenges. Ongoing research in gene therapy and innovative approaches is expected to expand the therapeutic landscape and improve patient outcomes globally by 2034.
Track the growth potential and innovation pipeline in the Tay-Sachs Disease Market with our detailed review.
Sandhoff Disease
Sandhoff Disease is a rare, inherited lysosomal storage disorder caused by mutations in the HEXB gene, resulting in a deficiency of both β-hexosaminidase A and B enzymes. This deficiency leads to the accumulation of GM2 gangliosides in neurons, causing progressive neurodegeneration and symptoms such as seizures, muscle weakness, vision and hearing loss, and developmental regression. Sandhoff Disease, inherited in an autosomal recessive manner, is clinically similar to Tay-Sachs Disease and presents in three forms: infantile, juvenile, and adult, with the infantile form being the most common and severe, often fatal by age 3 to 5.
As per secondary research, Sandhoff Disease is an uncommon condition affecting 1 in 1 million people worldwide. The National Institutes of Health confirms this ultra-rare prevalence, noting that the infantile form dominates, while juvenile and adult-onset forms are less frequent but increasingly identified through genetic screening. The global burden of Sandhoff Disease, though small, is significant due to its severity, with carrier screening in high-risk populations and improved diagnostics enhancing case detection, paving the way for earlier management of Sandhoff Disease symptoms.
There is currently no cure for Sandhoff Disease, and no FDA-approved drugs directly target the underlying enzyme deficiency. Treatment remains supportive and palliative, focusing on managing symptoms to improve quality of life. Anticonvulsant medications help control seizures, while nutritional support, such as gastrostomy tubes, ensures adequate hydration and nutrition for patients with swallowing difficulties. Physical therapy aids in maintaining mobility, and respiratory support may be required in advanced cases. These symptomatic treatments, critical to the Sandhoff Disease therapeutic landscape, do not halt the disease’s relentless progression, emphasizing the need for disease-modifying Sandhoff Disease drugs.
The pipeline for Sandhoff Disease includes promising research into gene therapy, enzyme replacement therapy, substrate reduction therapy, and stem cell-based approaches. TSHA-101, a gene therapy developed by Taysha Gene Therapies, is in early-stage clinical development, aiming to restore β-hexosaminidase activity and halt neurological decline, with preclinical studies showing encouraging results. Enzyme replacement therapy and substrate reduction therapy are under investigation to address the enzyme deficiency and reduce GM2 ganglioside buildup, respectively, though they remain preclinical. Stem cell therapies are also being explored. Companies like Bristol-Myers Squibb, Novartis, Pfizer, and Sanofi are among those contributing to the Sandhoff Disease pipeline, driven by orphan drug incentives. These emerging Sandhoff Disease therapies hold potential to reshape the treatment market, despite the challenges of an ultra-rare condition.
The Sandhoff Disease market remains small due to its ultra-rare nature but is gaining traction through orphan drug incentives, gene therapy advances, and rising awareness. DelveInsight projects growth in the 7MM, supported by improved vector manufacturing and strong collaborations among biotech, academia, and advocacy groups. Companies like Bristol-Myers Squibb Company, Catabasis Pharmaceuticals, Celgene Corporation, CRISPR Therapeutics AG, Genentech, and others are driving innovation. High unmet needs and potential per-patient treatment costs align with trends in other rare neurogenetic diseases. Emerging therapies like TSHA-101 offer hope for improved outcomes by 2034.
Explore opportunities and therapeutic advances in the Sandhoff Disease Market in our updated research.
Mucopolysaccharidoses
Mucopolysaccharidoses are a group of rare, inherited lysosomal storage disorders caused by deficiencies in enzymes that break down glycosaminoglycans, leading to their accumulation in organs and tissues. This buildup causes progressive damage, resulting in symptoms like skeletal deformities, organ enlargement, developmental delays, and vision or hearing loss. Mucopolysaccharidoses include several types, such as MPS I (Hurler, Scheie), MPS II (Hunter), MPS III (Sanfilippo), MPS IV (Morquio), MPS VI (Maroteaux-Lamy), MPS VII (Sly), and Type IX (Hyaluronidase Deficiency), each linked to a specific enzyme deficiency. These disorders vary in severity, with some causing severe neurological impairment and others primarily affecting physical function.
To help break down these complex conditions, we’ve created a dedicated blog that dives deep into the specific types of MPS, detailing their clinical features and genetic underpinnings. You can explore the full breakdown by reading the blog here
In the meantime, here’s a quick overview of a few key MPS types and how they differ in clinical presentation and treatment approaches.
MPS I (Hurler, Hurler-Scheie, and Scheie Syndromes)
MPS I is caused by alpha-L-iduronidase deficiency and presents in three forms—Hurler (severe), Hurler-Scheie (intermediate), and Scheie (mild). It affects various organs, with the severe form also impacting the CNS. As of 2024, there were around 660 diagnosed prevalent cases across the 7MM. ALDURAZYME (laronidase) is the approved ERT. Emerging therapies such as OTL-203, JR-171, RGX-111, and ISP-001 are currently in development, with a key focus on improving central nervous system (CNS) targeting. Valued at approximately USD 145 million in 2024, the MPS I market is projected to witness steady growth driven by these innovations. Click here to explore more about MPS I and its evolving treatment landscape.
MPS II (Hunter Syndrome)
Caused by iduronate-2-sulfatase deficiency, MPS II is X-linked and leads to progressive physical and neurological symptoms. While prevalence data for 2024 is limited, it is the second most common MPS in the 7MM. Approved treatments include ELAPRASE, HUNTERASE, and Japan’s IZCARGO, the first ERT to address CNS symptoms. Key pipeline therapies include DNL310 and SGT-003, targeting both peripheral and CNS manifestations.
MPS III (Sanfilippo Syndrome, Types A–D)
MPS III is a neurodegenerative disorder caused by impaired degradation of heparan sulfate, with four enzyme-deficient subtypes. It leads to early-onset behavioral and cognitive decline. There are no approved treatments yet, but gene therapies like UX111, DNL126, OTL-201, and LYS-SAF302 are in development, focusing on crossing the blood-brain barrier to slow neurological progression.
As research and innovation continue to advance, the landscape for Mucopolysaccharidoses treatment is evolving beyond traditional enzyme replacement therapies. With a growing pipeline of gene therapies and CNS-targeted treatments, there is renewed hope for improving outcomes and quality of life for patients and families affected by these devastating disorders. Stay tuned to our updates as we continue to track the latest breakthroughs and market developments in the MPS space.
Review strategic insights and future trends in the Mucopolysaccharidoses Market through our latest study.
Pompe Disease
Pompe Disease, or glycogen storage disease type II, is a rare lysosomal storage disorder caused by a deficiency of acid alpha-glucosidase, leading to glycogen buildup in muscles. This causes Pompe Disease symptoms like muscle weakness, respiratory issues, and heart problems, with severe infantile-onset cases often fatal early in life. Late-onset forms affect adults with milder symptoms.
In 2023, 8.6K individuals in the United States were diagnosed with Pompe Disease, with 98% being adults, per secondary research. Infantile-onset cases, comprising a significant portion, face high mortality due to severe Pompe Disease symptoms. About 80% of US infantile cases were CRIM-positive, indicating residual enzyme activity that affects treatment outcomes. The National Institutes of Health notes that newborn screening boosts early detection, particularly for late-onset forms, across the 7MM, supporting timely management of Pompe Disease symptoms and increasing diagnosed prevalence.
The FDA has approved enzyme replacement therapies for Pompe Disease, including MYOZYME and LUMIZYME (alglucosidase alfa) by Genzyme/Sanofi and POMBILITI (cipaglucosidase alfa-atga) by Amicus Therapeutics. These Pompe Disease drugs reduce glycogen buildup, easing muscle and respiratory symptoms, but limitations like infusion reactions persist. No cure exists, and supportive care, such as respiratory aids and physical therapy, is vital for managing Pompe Disease symptoms, particularly in infants, within the therapeutic landscape.
The Pompe Disease pipeline is limited, with ACTUS-101 (AAV2/8-LSPhGAA) by Asklepios Biopharmaceutical in Phase I/II gene therapy trials to restore enzyme activity. Rarity hinders trial recruitment, slowing progress. Other efforts explore next-generation enzyme replacement therapies, but the sparse pipeline underscores challenges in the Pompe Disease treatment market. Companies like Amicus and Sanofi remain active, aiming to address unmet needs with innovative Pompe Disease therapies.
In 2023, the Pompe Disease market across the 7MM was valued at USD 18.4 million, with the US holding 70% of the share. DelveInsight projects growth, driven by rising diagnoses, newborn screening, and therapies like ACTUS-101. Orphan drug incentives and advocacy fuel the Pompe Disease market, despite high costs. The therapeutic landscape is set to grow, promising better management of Pompe Disease symptoms by 2034.
Explore the Future of Pompe Disease Treatment and Market Growth in Our Latest Blog
Cystinosis
Cystinosis is a rare genetic disorder caused by CTNS gene mutations, leading to cystine accumulation in cells and damaging organs like the kidneys, eyes, and muscles. Cystinosis symptoms include kidney dysfunction, vision problems, and growth delays, with infantile, juvenile, and adult forms varying by onset. Infantile Cystinosis, the most severe, often requires early intervention.
In 2023, DelveInsight estimated 3K Cystinosis cases across the 7MM, with the US accounting for 44% of cases, or about 1,320 diagnoses. The National Institutes of Health highlights that newborn screening and genetic testing are improving early detection, particularly for infantile Cystinosis, across major markets. This enhances opportunities for the timely management of Cystinosis symptoms, especially kidney-related complications. The global burden, though rare, underscores the importance of advancing Cystinosis treatment to address organ damage and support patient outcomes.
The FDA has approved two cystine-reducing drugs for Cystinosis, which include PROCYSBI (delayed-release cysteamine bitartrate) and CYSTAGON (immediate-release cysteamine bitartrate), both by Horizon Therapeutics, which lower cystine levels to slow organ damage. For corneal cystine deposits, CYSTADROPS (cysteamine hydrochloride) eye drops by Recordati S.p.A. provide targeted therapy. These Cystinosis drugs are critical for managing symptoms and preserving kidney function, but no cure exists. Supportive care, like nutritional support, aids quality of life, forming the core of the Cystinosis therapeutic landscape.
The Cystinosis pipeline is limited but promising, with CRISPR/Cas9 gene editing exploring corrections to CTNS gene mutations to restore cystinosin function. This early-stage approach could offer a curative Cystinosis treatment by addressing the genetic root. No specific drug candidates are in late-stage trials, reflecting recruitment challenges for this rare disease. Research efforts by academic and biotech groups continue to advance the Cystinosis treatment market, aiming to transform the therapeutic landscape with innovative Cystinosis therapies.
In 2023, the Cystinosis market across the 7MM showed steady demand for treatments like PROCYSBI and CYSTADROPS. DelveInsight projects significant growth in the Cystinosis market, driven by rising diagnoses, improved screening, and potential gene therapies like CRISPR/Cas9. Orphan drug incentives and advocacy fuel the Cystinosis treatment market, despite high costs. The therapeutic landscape is poised to evolve, promising better management of Cystinosis symptoms and enhanced patient outcomes by 2034.
Understand trends and evolving treatment strategies in the Cystinosis Market with our in-depth analysis.
Danon Disease
Danon Disease is a rare X-linked genetic disorder caused by LAMP2 gene mutations, disrupting lysosomal function and autophagy. Primarily affecting males, it causes Danon Disease symptoms like hypertrophic cardiomyopathy, skeletal myopathy, and intellectual disability, with females showing milder, later-onset symptoms. Severe heart issues often lead to heart failure or sudden death in young males.
Due to its rarity, precise epidemiology for Danon Disease is limited. The National Institutes of Health suggests a prevalence of less than 1 in 1 million, with fewer than 1K diagnosed cases in the 7MM, based on rare disease registries. Males dominate severe cases, while females contribute to milder diagnoses. Improved genetic testing and cardiac screening are increasing detection of Danon Disease symptoms, particularly in families with a history of cardiomyopathy, supporting early intervention and highlighting the need for targeted Danon Disease treatment across major markets.
No FDA-approved drugs exist for Danon Disease, and treatment is supportive, focusing on symptom management. Beta-blockers and ACE inhibitors address cardiac issues, while implantable defibrillators prevent sudden death. Heart transplantation is a last resort for severe cases. Supportive care, like physical therapy for myopathy, is crucial in the Danon Disease therapeutic landscape. The absence of disease-modifying Danon Disease drugs underscores significant unmet needs, particularly for therapies that can prevent or slow cardiac deterioration and improve patient outcomes.
The Danon Disease pipeline includes early-stage research into enzyme replacement therapy and AAV-based gene therapy to restore LAMP2 function. Gene therapy, in preclinical and early clinical trials, shows promise for correcting the genetic defect, while ERT remains exploratory. The limited pipeline reflects challenges in recruiting patients for this ultra-rare condition. Biotech firms and academic groups are advancing these Danon Disease therapies, aiming to transform the Danon Disease treatment market with innovative approaches, though no late-stage candidates have yet been reported.
The Danon Disease market is nascent but growing, driven by high unmet needs and a rare disease research focus. DelveInsight projects expansion in the Danon Disease market, fueled by better diagnostics, increased awareness, and emerging therapies like gene therapy. Partnerships, funding, and regulatory support are accelerating the Danon Disease treatment market, despite challenges like high therapy costs and scalability. The therapeutic landscape is set to evolve, promising improved management of Danon Disease symptoms and better outcomes by 2034.
Access comprehensive insights into the Danon Disease Market, including pipeline developments and key players.
Wolman Disease
Wolman Disease, or lysosomal acid lipase deficiency, is a rare, autosomal recessive disorder caused by LIPA gene mutations, leading to deficient lysosomal acid lipase. This causes lipid buildup in organs like the liver, spleen, and intestines, triggering Wolman Disease symptoms such as hepatosplenomegaly, vomiting, and failure to thrive. The infantile form, appearing in early weeks, is often fatal within a year without treatment.
Epidemiology data for Wolman Disease is scarce due to its rarity. The National Institutes of Health suggests a prevalence of 1 in 350K, with fewer than 500 diagnosed cases across the 7MM, primarily infantile-onset. Improved genetic screening is increasing the detection of Wolman Disease symptoms, particularly in newborns, enabling earlier intervention. The global burden, though small, drives research to address this life-threatening condition, with diagnostic advances supporting the need for accessible Wolman Disease treatment in major markets.
The FDA approved KANUMA (sebelipase alfa) in 2015, a recombinant enzyme replacement therapy by Alexion Pharmaceuticals, as the only drug for Wolman Disease. KANUMA reduces lipid accumulation, shrinks liver size, and extends survival, but challenges like immunogenic reactions and high-cost infusions persist. No cure exists, and supportive care, including nutritional support and symptom management, remains vital in the Wolman Disease therapeutic landscape. KANUMA’s approval marked a significant advance, yet unmet needs for more accessible Wolman Disease drugs continue.
The Wolman Disease pipeline is limited but promising, with AAV-mediated gene therapy in preclinical and early clinical stages to restore lysosomal acid lipase function. Hematopoietic stem cell transplantation is also under investigation, though experimental. Innovations in enzyme replacement therapy aim to reduce immunogenicity and dosing frequency. These emerging Wolman Disease therapies, driven by biotech and academic research, face recruitment challenges due to rarity but hold potential to transform the Wolman Disease treatment market.
The Wolman Disease market is small but growing, driven by KANUMA’s impact and high unmet needs. DelveInsight projects expansion from 2025 to 2034, fueled by diagnostic improvements, gene therapy advances, and partnerships between biotech and advocacy groups. Orphan drug incentives support the Wolman Disease market, despite high costs. The therapeutic landscape is evolving, with innovative Wolman Disease therapies promising better symptom management and improved infant outcomes by 2034.
Explore a complete market overview and future prospects in the Wolman Disease Market in our latest report.

What Does the Future Hold?
Lysosomal Storage Disorders may be rare, but their impact is profound, silently disrupting lives and stretching the limits of medical care. Yet, we now stand at the cusp of a scientific revolution. With cutting-edge gene therapies, next-generation enzyme replacements, and precision diagnostics advancing rapidly, the future of LSD care is no longer just about managing symptoms, it is about rewriting the narrative.
What was once a field plagued by limited options and high mortality is now evolving into one of the most promising frontiers in rare disease innovation. Unprecedented investments, regulatory incentives, and global advocacy are aligning to accelerate breakthroughs. Biotech pioneers, clinicians, and researchers are driving momentum toward durable, possibly curative solutions.
As we look towards the future, the LSD market is poised to shift from incremental change to transformative progress, offering renewed hope, extended lifespans, and improved quality of life for patients across the globe. The future is not just hopeful, it is within reach.

Downloads
Article in PDF
Recent Articles
- Bubble Boy Syndrome (ADA-SCID) – A Rare Immunodeficiency Disorder
- Exploring the Current Alzheimer’s Disease Drug Development Pipeline
- Breakthrough Device Designation granted to the AI Software for CTEPH
- Lupin acquired; FDA grants approval; NATCO gets approval; Wockhardt Ltd. gets import alert; Paten...
- Hepatic Encephalopathy- An ailment of liver to brain



