The Evolving Neurofibromatosis Treatment Landscape
Dec 27, 2021
Table of Contents
Neurofibromatosis is a progressive and autosomal dominant inherited disorder of the nervous system which causes tumors to form on the nerves anywhere in the body, at any time. Neurofibromatosis skin lesions are typically flat, pigmented patches but occasionally are elevated flesh-colored bumps.
There are three diferent Neurofibromatosis types:
Downloads
Click Here To Get the Article in PDF

Recent Articles
Type 1 Neurofibromatosis (NF1)
It causes skin changes and deformed bones can affect the spinal cord and brain, often contributing to learning disabilities, and usually starts at birth. NF1 (also known as von Recklinghausen’s disease) is a genetic tumor-disposition syndrome associated with malformations and tumor growth in the nervous system and other organ systems. Its frequency is approximately 1 per 3000 neonates, regardless of sex or ethnic background. Neurofibromatosis Type 1 symptoms include skin, bone, and eye abnormalities, nerve tumors (neurofibromas), learning and attention deficits and heart defects. The clinical manifestations for NF1 account for neurofibroma & other musculoskeletal manifestations.
The diagnosis of Type 1 Neurofibromatosis is made through clinical assessment including patient history and physical examination. The diagnosis is made on several factors such as six or more Neurofibromatosis cafe au laite macules > 5 mm in prepubertal individuals and > 15 mm in diameter in adults, two or more neurofibromas, freckling in the axillary or inguinal regions, two or more lisch nodules, abnormal development of the spine (scoliosis), the sphenoid bone, or the tibia.
Type 2 Neurofibromatosis (NF2)
It causes hearing loss, ringing in the ears, and poor balance. Type 2 Neurofibromatosis is a tumor disposition syndrome of autosomal dominant inheritance that is found in approximately 1 per 33,000 births. Half of all cases arise through de novo mutations. First described in 1882, it is the most common type of Neurofibromatosis and accounts for 90% of all cases. Neurofibromatosis Type 2 symptoms include nervous system tumors, cataracts, tinnitus (ringing or buzzing in the ears), hearing loss, and balance problems. Several clinical manifestations of NF2 include vestibular nerve schwannoma, meningiomas, spinal tumors, ependymomas and peripheral neuropathy.
There are a number of Neurofibromatosis testing and screening methods for the diagnosis of Neurofibromatosis Type 2 such as physical examination, medical history and tests for particular symptoms, such as hearing or balance tests.
Schwannomatosis is the rarest Neurofibromatosis form and causes intense pain. Schwannomatosis is genetically and clinically distinct from NF1 and NF2. In many cases, mutation of the SMARCB or LZTR1 genes is associated with the disease; however, the genetic cause of SWN in some people is unknown. Chronic pain is generally the most prominent symptom and is present anywhere on the body. This pain may or may not be associated with a specific schwannoma. Other symptoms include numbness, tingling, or weakness in the fingers and toes and/or loss of muscle function. Clinical manifestations of Schwannomatosis include factors such as schwannomas, pain, and other tumors.
The diagnosis includes identification of Schwannomatosis is defined by the occurrence of multiple schwannomas without any other stigmata of Type 2 Neurofibromatosis (bilateral vestibular nerve schwannomas, subcapsular cataract, NF2 mutation in the blood).
NF1 and NF2 are inherited in an autosomal dominant pattern, with an approximately 50% rate of de novo mutations in each. The genetic mechanism of SWN is more complex and the rate of new mutations probably lies between 50% and 80%.
Neurofibromatosis treatment of the tumors depends on the size, location, and symptoms of the tumors. Small tumors in patients with no symptoms can often be observed over time with yearly MR. For larger or symptomatic tumors, or tumors showing growth over time, surgical removal is usually recommended.
DelveInsight estimates indicate that incident cases of Type 1 Neurofibromatosis were observed to be 1 in every 3,000 children born. Whereas incident cases of Type 2 Neurofibromatosis occurred 1 in every 25,000 people worldwide, it is considered to be more rare. In addition to this the third type, incident cases of Schwannomatosis were reported to be 1 in every 40,000 people.
Neurofibromatosis Treatment Outlook
Neurofibromatosis treatment depends on the presence and severity of the forearm, lower extremity, or spinal deformity. People with Type 1 Neurofibromatosis do not usually require any prolonged treatment for any manifestation (disease signs or development) during their lives. Surgery is frequently impossible but may be required to improve or salvage nerve function, after considering the cost-benefit ratio. Cutaneous neurofibromas can be surgically resected for either esthetic or medical reasons. For malignant peripheral nerve sheath tumors, complete surgical resection is the single treatment approach offering a chance of cure; the 5-year survival rate with chemotherapy is less than 20%.
Neurofibromatosis type 1 treatment for other conditions is aimed at controlling or relieving symptoms. Headache and seizures are treated with medications. Vitamin D3 deficiency, if present, should be substituted. Genetic counseling is especially important. Cognitive deficits and developmental delays should be recognized early and addressed with suitable special-needs education.
Type 2 Neurofibromatosis medication includes an off-label treatment with the vascular endothelial growth factor (VEGF) inhibitor – bevacizumab that has now become an important therapeutic option. At least over the intermediate-term, it is associated with hearing improvement in 57% and tumor shrinkage in 55% of patients, although there is a risk of side effects including hypertension and proteinuria. Apart from that surgery is always the last treatment option.
Schwannomatosis treatment involves functionally limiting schwannomas that can be surgically resected. Genetic investigation of the resected tumor material is of high prognostic relevance, in particular when the diagnosis is initially unclear. Co-analgetic drugs (amitriptyline, gabapentin, pregabalin) have been found especially useful in Neurofibromatosis pain management associated with neuropathic pain.
In April 2020, the US Food and Drug Administration approved selumetinib (Koselugo) as a treatment for Neurofibromatosis in children aged 2 years and older.
Current Marketed Neurofibromatosis Treatment Drugs
People with Neurofibromatosis are generally monitored periodically for the development of associated tumors and other health problems. Neurofibromatosis treatment depends on the signs and symptoms present in each person. For example, vestibular schwannomas are typically slow-growing tumors so they may be observed for a period of time before treatment becomes necessary. When they begin to cause symptoms, vestibular schwannomas are usually treated with surgery and less frequently, with radiation therapy.
Other tumors associated with Neurofibromatosis are also treated surgically, although Antineoplastic Agents, Corticosteroids, Antihistamines may also be recommended in rare circumstances. Antineoplastic agents such as vincristine, cisplatin and lomustine act by inhibiting the key factors responsible for the neoplastic transformation of cells. Corticosteroids such as prednisone have anti-inflammatory properties and cause profound and varied metabolic effects. Antihistamines such as diphenhydramine may control itching by blocking the effects of endogenously released histamine.
There is no permanent cure for Neurofibromatosis and at present no other effective medical therapy apart from selumetinib is known for Neurofibromatosis. Also, the treatment does not assure a complete cure and involves constant medication throughout the therapeutic course. There are certain shortcomings associated with complete surgical resection. Surgeries can completely remove tumors but always have chances of nerve damage, whereas focused radiation therapy effectively reduces tumor size and minimizes the risk of acquiring paralysis.
Pharmaceutical companies such as AstraZeneca, Array Biopharma, SpringWorks Therapeutics, Novartis, Recursion Pharmaceuticals, etc. are actively working towards the development of potential therapies in order to fulfill the unmet medical needs of the currently used therapeutics.

Analysis of Emerging Drugs in Neurofibromatosis Treatment
The dynamics of the Neurofibromatosis market are anticipated to change in the coming years owing to the improvement in the diagnosis methodologies, rising awareness of the diseases, incremental healthcare spending across the world and the expected launch of emerging therapies. Companies across the globe are diligently working towards the development of new treatment therapies for Neurofibromatosis. This includes some of the key players such as Neurofibromatosis are AstraZeneca, Array Biopharma, SpringWorks Therapeutics, etc. The launch of emerging therapies such Selumetinib (AstraZeneca), Binimetinib (Array Biopharma/ Novartis) and PD-0325901 (SpringWorks Therapeutics), Trametinib (Novartis), will significantly impact the Neurofibromatosis market in the coming years.
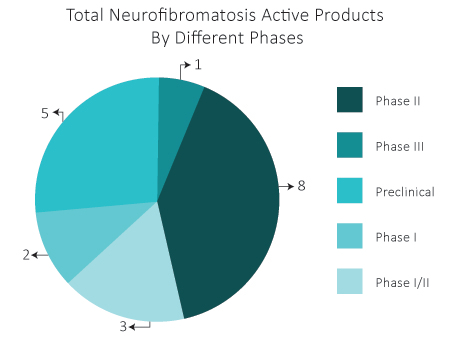
As per DelveInsight estimates, in the Neurofibromatosis therapeutics pipeline, there are almost 19 drugs that are in different phases of development.
PD-0325901 by SpringWorks Therapeutics is an investigational oral, small molecule, MEK1 and MEK2 inhibitor that has been tested in Phase I and Phase II clinical trials. The U.S. Food and Drug Administration (FDA) has granted Orphan Drug designation for PD-0325901 for Neurofibromatosis Type 1 treatment. SpringWorks has initiated a Phase IIb study of PD-0325901 in patients with NF1-associated plexiform neurofibromas (PN).
Binimetinib is being developed by Array Biopharma and currently, it is in the Phase II stage of development for the treatment of Children and Adults with NF1 Associated Plexiform Neurofibromas. In June 2019, Array Biopharma announced they have entered into a definitive merger agreement under which Pfizer will acquire Array. Pfizer has agreed to acquire Array for a total enterprise value of approximately $11.4 billion.
Trametinib is being developed by Novartis and is a reversible inhibitor of mitogen-activated extracellular signal-regulated kinase 1 (MEK1), MEK2 activation and of MEK1 and MEK2 kinase activity developed by Novartis for the treatment of unresectable or metastatic melanoma with BRAF V600E. This drug is currently in Phase II for treatment of NF1-related plexiform neurofibroma with trametinib (plexifpc).
Selumetinib by AstraZeneca is an orally active, small molecule with potential antineoplastic activity. Selumetinib is an ATP-independent inhibitor of mitogen-activated protein kinase 1 and 2. Inhibition of both MEK1 and 2 by selumetinib prevents the activation of MEK1/2 dependent effector proteins and transcription factors, thereby leading to an inhibition of cellular proliferation in various cancers. This drug is currently in Phase II and the trial is expected to be completed by May 2022 with 34 enrolled participants.
REC-2282 is being developed by Recursion Pharmaceuticals as an orally bioavailable, CNS-penetrating, pan-histone deacetylase, or HDAC, inhibitor with PI3K/AKT/mTOR pathway modulatory activity. By comparison to the marketed HDAC inhibitors, REC-2282 is uniquely suited for patients with NF2, and NF2-mutant CNS tumors, due to its oral bioavailability and CNS exposure. The company expects to enroll the first patient in an adaptive, parallel-group, Phase II/III, randomized, multicenter study within the next four to five quarters.
Pexidartinib/PLX3397 is being developedby Plexxikon in collaboration with National Cancer Institute (NCI), they initiated a Phase I/II Trial of PLX3397 in Children and Young Adults with Refractory Leukemias and Refractory Solid Tumors Including Neurofibromatosis Type 1 (NF1) Associated Plexiform Neurofibromas (PN). Pexidartinib is an orally bioavailable small-molecule tyrosine kinase inhibitor that inhibits colony-stimulating factor-1 receptor (CSF-1R).
Unmet Needs in Neurofibromatosis Treatment Landscape
Need for novel therapies
There is an increased rate of malignant peripheral nerve sheath tumors and this type of sarcoma develops in patients with Neurofibromatosis Type 1. These tumors are highly aggressive, very difficult to treat, and don’t respond well to traditional chemotherapy or radiation. There is an urgent need for better therapies for the treatment of malignant peripheral nerve sheath tumors as well as other cancer types that can be associated with Neurofibromatosis. A large proportion of children diagnosed with Neurofibromatosis type 1-related plexiform neurofibromas have no appropriate treatment available to them and represent a significant unmet medical need.
Need for new approaches to Neurofibromatosis treatment
Given the potential for major morbidity and even mortality, a strong impetus exists to develop new approaches to treatment. The complexity and variability of the phenotypes observed in people with Neurofibromatosis present challenges that require careful consideration before any kind of ‘cure’ can be considered. Selumetinib is the first drug that’s been proven to have efficacy, there is a need of newer agents with increased efficacy and activity with reduced toxicity and more durable responses. This would mean third- and fourth-generation MEK inhibitors as well as other types of signal transduction inhibitors that can slow down tumor growth.
Disease burden and reduced quality of life
In particular, cutaneous neurofibromas—which are neurofibromas on the skin—can be very disfiguring and affect patients’ quality of life (QOL). They are not as medically harmful as the plexiform neurofibromas but have a tremendous impact on patients in terms of their social lives and have a wide array of effects on QOL for these patients. Additionally, problems with learning, attention, and anxiety are very common and prevalent in this population and similarly can have a large impact on the lives of our patients. The severe burden of Neurofibromatosis with plexiform neurofibromas on the QOL of patients demonstrates the high unmet need for an effective treatment option that can reduce tumor burden and improve QOL.

FAQs
The first and most common method is a clinical diagnosis as the signs of NF1 will be observed in the patient’s skin, eyes, bones or brain.
The second method is genetic testing, also called a molecular or DNA diagnosis which requires a blood sample.
If there are no complications, Neurofibromatosis life expectancy is almost normal. With the right education, people with Neurofibromatosis can live a normal life.
Neurofibromatosis is a rare disorder that affects males and females in equal numbers. It affects all races and ethnic groups equally.
Unfortunately, there is no existent cure for Neurofibromatosis, but signs and symptoms can be managed given proper treatment is provided.
Downloads
Article in PDF



