The Race to Transform Charcot-Marie-Tooth Disease Treatment: Which Therapy Will Cross the Finish Line First?
Jul 10, 2026
Table of Contents
Summary
- Charcot-Marie-Tooth disease is genetically heterogeneous, with more than 100 genes associated with different CMT disease subtypes.
- Currently, no approved therapies are available for the treatment of CMT disease, leaving patients with limited treatment options. As a result, there remains a significant unmet need for effective therapies that can modify disease progression rather than simply manage symptoms.
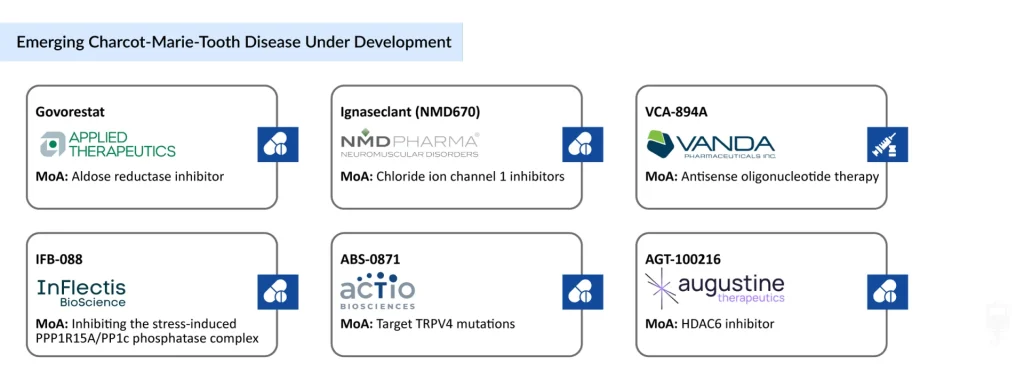
- Some of the CMT disease drugs under development include IFB-088 (InFlectis BioSciences), Govorestat (Applied Therapeutics), Ignaseclant (NMD670) (NMD Pharma), ABS-0871 (Actio Biosciences), VCA-894A (Vanda Pharmaceuticals), EDK060 (Novartis), AGT-100216 (Augustine Therapeutics), and others.
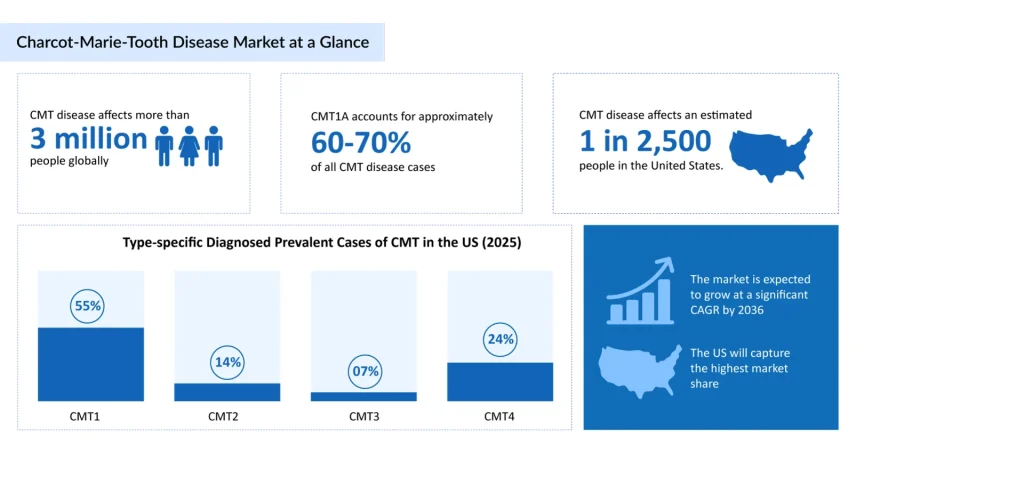
Charcot-Marie-Tooth disease is often labeled “rare,” but the numbers tell a more nuanced story. Charcot-Marie-Tooth disease is considered to be one of the most common of the 7,000 rare diseases affecting children. According to the Hereditary Neuropathy Foundation, CMT affects an estimated 1 in 2,500 people in the US, translating to roughly 2.6 million people affected worldwide, and many experts believe the true figure is higher due to underdiagnosis. The Muscular Dystrophy Association estimates a global CMT prevalence of approximately 19 cases per 100,000 people, while the National Organization for Rare Disorders places worldwide prevalence in a broader range of 9–28 per 100,000. The Charcot-Marie-Tooth Association puts the global patient population at more than 3 million. Regional data adds further texture: incidence is reported at roughly 1 in 2,500 in Europe and the US, compared with about 1 in 10,000 in Japan, and as high as 1 in 1,500 in parts of France.
Downloads
Click Here To Get the Article in PDF

Recent Articles
- FDA Expands SOLIRIS for Pediatric Myasthenia Gravis; vTv’s Cadisegliatin Program Resumes as FDA L...
- Chimerix Submits Dordaviprone NDA for Accelerated Approval in Recurrent H3 K27M-Mutant Diffuse Gl...
- Neurogene raises $68.5M; AbbVie and Teneobio sign deal; 5AM Ventures nets
- FDA Fast Track Status to Kyverna’s KYV-101; Annovis’s Phase III Study for Buntanetap; Gilteritini...
Among subtypes, Charcot-Marie-Tooth disease type 1A is the most frequently diagnosed, caused by a duplication on chromosome 17p11.2 that leads to overexpression of the PMP22 gene, accounting for roughly 55–70% of all CMT cases, depending on the population studied. Charcot-Marie-Tooth disease type 2 and X-linked CMT (CMTX1) make up most of the remainder, with type 2 subtypes typically presenting with an axonal (rather than demyelinating) pattern of nerve damage and, in several forms, a later average age of onset.
The patient burden extends well beyond the nerves. People living with CMT disease often cope with progressive foot deformities, chronic pain, fatigue, and a real risk of disability, including loss of independent ambulation in advanced cases. Because there is currently no approved disease-modifying therapy, the day-to-day burden, orthotics, bracing, repeated corrective surgeries, mobility aids, and lost work productivity, falls heavily on patients and caregivers alike.
The Current Reality of CMT Treatment
Here’s the sobering reality that shapes the entire CMT disease treatment landscape: there is currently no FDA-approved, disease-modifying drug for Charcot-Marie-Tooth disease. Every existing option is symptomatic and supportive rather than curative, aimed at managing pain, preserving function, and slowing the practical impact of nerve degeneration rather than reversing it. Charcot-Marie-Tooth disease physiotherapy sits at the center of this approach, a structured, physician-guided exercise regimen combining muscle-strength training, stretching to prevent contractures, and moderate aerobic conditioning. Started early, physical therapy treatment for Charcot-Marie-Tooth disease can meaningfully delay the progression of muscle weakness and help patients maintain stamina, balance, and everyday function for longer. Occupational therapy complements this by teaching adapted techniques for daily living tasks, while orthotic devices, ankle-foot orthoses, custom shoe inserts, and leg braces help correct gait abnormalities and reduce fall risk. In more advanced cases, corrective surgery may be used to address severe foot deformities like high arches or hammertoes.
On the pharmacological side, treatment is limited almost entirely to symptom control. Non-opioid and opioid analgesics are used to manage musculoskeletal pain arising from the mechanical stresses CMT places on joints, while neuropathic pain, caused by the nerve damage itself, is often managed with tricyclic antidepressants or anticonvulsant medications. Genetic counseling rounds out the current standard of care, helping patients and families understand recurrence risk, connect with support and research communities, and plan proactively. It’s a comprehensive but fundamentally reactive model of care, which is exactly why the emerging therapeutic pipeline described below represents such a meaningful shift for the CMT community.
The CMT Pipeline is Finally Catching Up to a Long-Ignored Unmet Need
The good news for a disease with no approved disease-modifying option: the CMT pipeline is finally catching up to the unmet need, with several promising drug candidates advancing through clinical development. Several biopharmaceutical companies are actively advancing disease-modifying therapies for Charcot-Marie-Tooth disease, reflecting the substantial unmet medical need in this rare inherited neuropathy.
Among the leading clinical-stage programs, InFlectis BioScience is developing IFB-088, a small molecule designed to restore myelination and improve peripheral nerve function. Applied Therapeutics is evaluating govorestat, an aldose reductase inhibitor that targets metabolic pathways associated with CMT, while NMD Pharma is advancing NMD670, a neuromuscular junction modulator intended to improve muscle strength and motor performance. Meanwhile, Novartis is developing EDK-060, a gene therapy candidate aimed at correcting the underlying genetic defects responsible for specific CMT subtypes.
Another promising investigational therapy is ABS-0871 from Actio Biosciences, a TRPV4 inhibitor currently in Phase I clinical development. Preclinical studies in CMT2C models have demonstrated improvements in motor function and mobility, and the program has received both FDA Orphan Drug and Rare Pediatric Disease designations, highlighting its potential to address a critical unmet need.
The early-stage pipeline continues to expand with innovative precision medicine approaches. Vanda Pharmaceuticals is developing VCA-894A, an antisense oligonucleotide (ASO) therapy targeting a cryptic splice-site variant in the IGHMBP2 gene responsible for CMT2S, an ultra-rare pediatric-onset axonal subtype. Following the first patient dosing in its Phase I trial in June 2025, the therapy received FDA Rare Pediatric Disease Designation in July 2026, making it eligible for a Priority Review Voucher upon potential approval and reinforcing the growing regulatory support for genotype-specific therapies in CMT. Likewise, Augustine Therapeutics has initiated Phase I evaluation of AGT-100216, further broadening the pipeline of next-generation CMT therapies.
Beyond the seven major markets, several companies are also pursuing novel treatment strategies. ENCell is developing EN001, an allogeneic Wharton’s jelly-derived mesenchymal stem cell therapy, as a potential regenerative treatment for CMT. In March 2025, the investigational therapy received FDA Orphan Drug Designation, underscoring its promise for patients with this rare disorder.
Collectively, the CMT drug pipeline is evolving from symptomatic management toward disease-modifying and genetically targeted interventions. Advances across small molecules, gene therapies, ion channel modulators, stem cell therapies, and precision antisense oligonucleotides highlight a rapidly expanding therapeutic landscape with the potential to transform the future treatment paradigm for Charcot-Marie-Tooth disease.
Future Outlook of CMT Treatment Landscape
The Charcot-Marie-Tooth disease treatment landscape is standing at a genuine inflection point. For decades, care has been built almost entirely around bracing, physiotherapy, and pain management, necessary but incapable of altering the underlying course of the disease. That is starting to change. With multiple programs now in Phase I and Phase II development, spanning distinct mechanisms and even distinct genetic subtypes like CMT-SORD, CMT2C, and CMT2S, the field is moving toward a future where treatment can be matched to a patient’s specific genetic profile rather than applied as a one-size-fits-all symptomatic regimen.
Regulatory momentum is reinforcing this shift. Orphan Drug Designations, Rare Pediatric Disease Designations, and active FDA engagement, including Applied Therapeutics’ recent Type C meeting on govorestat and Vanda’s newly granted pediatric designation for VCA-894A, all signal that agencies recognize the urgency of the unmet need in CMT. Advances in genetic testing and neurophysiological diagnostics are also expected to shorten the historically long road to diagnosis, enabling earlier intervention precisely when emerging therapies are likely to be most effective at slowing nerve degeneration.
The CMT field is evolving rapidly, with significant advances in genetic diagnosis and disease recognition, as per Sadaf Javed, Functional Head of Forecasting at DelveInsight. International networks and patient organization partnerships are vital for progress, enabling collaboration and large-scale studies, Javed further added. Gene therapy, Javed said, shows promise but currently faces safety and targeting challenges; PMP22 silencers for CMT1A are close to being tested in patients.
Challenges remain, of course. The sheer genetic heterogeneity of CMT, over 100 implicated genes, complicates both clinical trial design and eventual treatment standardization, and past setbacks, such as the Phase III PREMIER trial failure for PXT3003 in CMT1A, are a reminder that the path to approval is neither short nor guaranteed. Even so, with a strengthening pipeline, growing patient advocacy, and regulatory bodies actively clearing pathways for accelerated development, the next decade looks poised to deliver what CMT patients have waited over a century for: real, disease-modifying treatment options.

FAQs
In the simplest terms, the definition of Charcot-Marie-Tooth disease is this: it’s a group of genetically diverse disorders that damage the peripheral nerves, the nerves outside the brain and spinal cord that carry motor and sensory signals to the arms, hands, legs, and feet. Unlike muscular dystrophy, which directly attacks muscle tissue, CMT disease meaning centers on the nerves themselves; the muscles weaken and waste away only as a secondary consequence of losing their nerve signal. This is an important distinction, and it’s one reason people frequently search for muscular dystrophy and Charcot-Marie-Tooth disease together, the two conditions share overlapping symptoms but very different underlying biology. Although there is no approved disease-modifying therapy yet, treatment advances include gene-targeted therapies, RNA-based approaches, and novel small molecules currently in clinical development, alongside supportive care such as physical therapy, orthotics, and surgery to improve mobility and quality of life.
The name honors the three physicians, Jean-Martin Charcot, Pierre Marie, and Howard Henry Tooth, who independently described the condition in 1886. More than a century later, CMT disease, or Charcot-Marie-Tooth disease, remains the umbrella term for what scientists now recognize as over 100 different genetic subtypes.
So, what is the cause of Charcot-Marie-Tooth disease at the molecular level? CMT disease causes trace back to mutations, duplications, or deletions in any of more than 100 identified genes responsible for building and maintaining the peripheral nerves’ insulating myelin sheath or their internal axonal machinery. A single gene, PMP22, when duplicated, accounts for around half of all cases. In roughly 40% of cases, however, no responsible gene has yet been pinpointed, which is precisely why Charcot-Marie-Tooth disease testing and expanded genetic panels remain such an active area of clinical research.
This is one of the most searched and most reassuring questions patients and families ask. In the vast majority of cases, Charcot-Marie-Tooth disease is not considered fatal, and life expectancy is generally normal. So, can you die from Charcot-Marie-Tooth disease directly? Typically, no CMT disease death is rare and is not a direct outcome of the condition itself for most patients. That said, a small number of rare and severe subtypes can involve breathing muscle involvement or overlapping complications that require close monitoring, so “is Charcot-Marie-Tooth disease fatal” isn’t a one-size-fits-all answer; it depends heavily on the specific genetic subtype and severity.
The growth of the Charcot-Marie-Tooth disease market is being driven by rising disease awareness, advances in genetic testing enabling earlier diagnosis, and a robust pipeline of gene therapies and disease-modifying treatments. Additionally, rising investments in rare disease research, supportive regulatory incentives, and growing interest from biotechnology and pharmaceutical companies are accelerating therapeutic development and market expansion.
Leading companies such as InFlectis BioScience, Applied Therapeutics, NMD Pharma, Novartis, Actio Biosciences, Vanda Pharmaceuticals, Augustine Therapeutics, ENCell, and others are currently evaluating their lead assets in various stages of development.
Some of the CMT disease drugs under development include IFB-088, govorestat, NMD670, EDK-060, ABS-0871, VCA-894A, AGT-100216, EN001, and others.
DelveInsight’s Charcot-Marie-Tooth Disease Market report provides a comprehensive analysis of the current and future market landscape, including disease epidemiology, market size forecasts, emerging therapies, and key companies developing novel treatments. It also highlights market drivers, barriers, unmet needs, competitive dynamics, and commercialization opportunities across the major markets, enabling stakeholders to make informed strategic decisions.
Downloads
Article in PDF
Recent Articles
- FDA Fast Track Status to Kyverna’s KYV-101; Annovis’s Phase III Study for Buntanetap; Gilteritini...
- Chimerix Submits Dordaviprone NDA for Accelerated Approval in Recurrent H3 K27M-Mutant Diffuse Gl...
- FDA Expands SOLIRIS for Pediatric Myasthenia Gravis; vTv’s Cadisegliatin Program Resumes as FDA L...
- Neurogene raises $68.5M; AbbVie and Teneobio sign deal; 5AM Ventures nets



