Retinitis Pigmentosa: A complex disease, and the increasing gap between prevalence and diagnosed prevalence
Mar 23, 2020
Table of Contents
DelveInsight’s Retinitis Pigmentosa Epidemiology forecast analysis revealed that the prevalent population of the disease is expected to increase during the study period 2017-30.
Retinitis Pigmentosa Defects and Diagnosis
A group of inherited genetic retinal diseases – Retinitis pigmentosa – leads to progressive deterioration of the ability of eyes to see. The name was first used by Dr Franciscus Donder, a Dutch ophthalmologist, in the year 1857.
Downloads
Click Here To Get the Article in PDF

Recent Articles
- Merck’s Sotatercept Trial Result; PARP Rivals Closing in on AstraZeneca and Merck’s Lynparza; FDA...
- FDA Approves LUMAKRAS with VECTIBIX for KRAS G12C-Mutated Colorectal Cancer; PYC Receives FDA Rar...
- Actinium Announces SIERRA Trial Results; Santhera Seeks FDA Review for Vamorolone; Seres Announce...
- Merck’s Keytruda Wins Another FDA Approval; Sanofi Pauses Trial of Myasthenia Gravis Drug, tolebr...
- The Hidden World of Vision: Exploring the Top 5 Rare Eye Diseases
The defect inflicted upon the light-sensitive tissue – the retina – of the eye, Retinitis pigmentosa is the main cause of inherited form of blindness.
First and foremost, the sign of Retinitis pigmentosa is usually a loss in the ability to see during the night, which becomes apparent in the early childhood of a person. The disease progresses with the formation of blind spots in the peripheral vision, which further merges to become tunnel vision, eventually leading to loss of vision.
Retinitis – is an inflammation of the retina; however, the condition Retinitis pigmentosa does not involve any type of inflammation of the eye. Hence, many claims that here the term defies the meaning of the disorder. The main reason behind the diseases involves genetic mutations in any one of more than 50 genes – genes that are responsible for transcribing proteins necessary
In some mutations, there is a dearth of the protein required, in some, there is a production of a toxic protein, and in remaining cases, there is a presence of abnormal protein, which is good for nothing. The result of mutation might differ; however, the result is the same – damaged photoreceptors.
Usually, during Retinitis pigmentosa diagnosis and physical examination of the retina through an ophthalmoscope, dark pigmentation – spots are revealed on the retina. Other tests are also in the order for Retinitis pigmentosa diagnosis such as Genetic testing – for confirmation of inheritance pattern, Electroretinography, Visual field testing, and Optimal coherence tomography.
Retinitis Pigmentosa Epidemiology Forecast and Analysis
Being a rare disease, gathering and evaluating Retinitis Pigmentosa epidemiological data is rather a challenging task, and the data is often available in fragments and partial form.
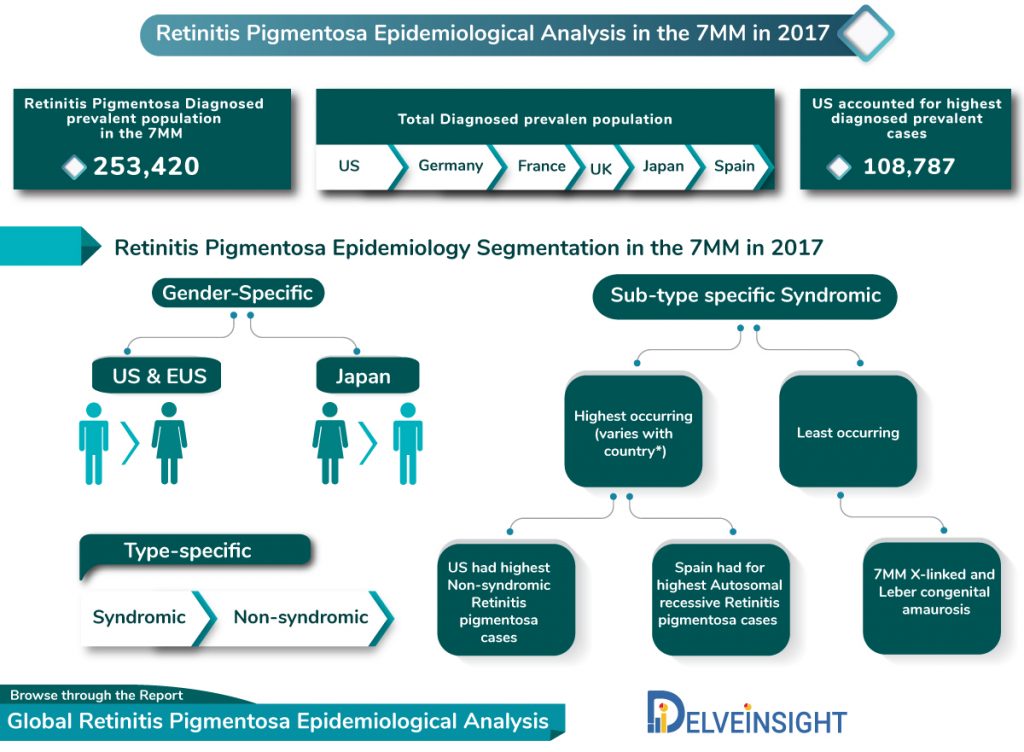
As per DelveInsight’s Retinitis pigmentosa, epidemiological model, the diagnosed Retinitis Pigmentosa prevalent population in the 7MM (the US, EU5 and Japan) was observed to be 253,420 in 2017. The most diagnosed cases of the disease were observed in the US, around 108,787. In EU5, Germany recorded the highest diagnosed Retinitis Pigmentosa prevalent population with 30,642 cases, followed by France and the UK. Spain was observed with least Retinitis pigmentosa prevalent population, with Japan reported being around 28,051 Retinitis pigmentosa prevalent cases.
As per DelveInsight’s Retinitis Pigmentosa Epidemiology Forecast analysis, the prevalence of the disease is expected to rise in the coming decade in the 7MM.
Analysis of Retinitis pigmentosa epidemiological data is segmented based on various further factors such as the prevalence in males as compared to females, types of Retinitis pigmentosa, and further sub-types of types of the disease as described below.
Retinitis Pigmentosa Gender-specific Epidemiology
A higher preponderance of Retinitis pigmentosa is observed in males, as compared to females in the US and EU5 in 2017 However; the case reverses, in case of Japan, where females form a large pool of Retinitis pigmentosa prevalent population, estimated DelveInsight’s Retinitis pigmentosa epidemiological analysis and forecast.
Retinitis Pigmentosa type-specific epidemiology
Retinitis pigmentosa can be categorized into two major sub-types – Syndromic Retinitis Pigmentosa and Non-syndromic Retinitis Pigmentosa.
Majority of the patients get diagnosed with non-syndromic Retinitis pigmentosa, which in 2017 was recorded to be 172,256 cases in the 7MM.
However; if the disease is present with symptoms such as photopsia (sensation of lights flashing), abnormal central vision, abnormal colour vision, or marked asymmetry in ocular involvement, then it is not necessary the reason behind is Retinitis pigmentosa. The patient may have other retinal degeneration disorders, such as Usher syndrome, and other genetic syndromes, including Bardet-Biedl syndrome; Refsum disease; and neuropathy, ataxia, and retinitis pigmentosa (NARP), Retinitis pigmentosa do sometimes occur in syndromic form.
They are considered in the differential diagnosis of typical retinitis pigmentosa (RP).
In the 7MM, only one-fourth of the total diagnoses Retinitis pigmentosa prevalent population were observed to be suffering from a syndromic form of the disease.
Among the Syndromic Retinitis pigmentosa cases Usher Syndrome and Bardet-Biedl syndrome are the most diagnosed prevalent subtypes of the disease. Whereas Non-syndromic Retinitis pigmentosa cases presented a varied picture in the 7MM, for some geographies like in the US, it was sporadic non-syndromic Retinitis pigmentosa with are the highest contribution, for others like in Spain Autosomal recessive Retinitis Pigmentosa accounted for the maximum cases. However, in all the 7MM countries, X-linked RP and Leber congenital amaurosis remain the least occurring subtypes of the disease
Way forward – Retinitis pigmentosa Epidemiological forecast
The overall management of Retinitis pigmentosa or any rare disease is quite difficult due to several issues including epidemiological ones. Non-availability of data or availability in an unorganized manner is not one of the major Retinitis pigmentosa market barriers. For any epidemiological model to present accurate values, quantitative estimation of the disease and adequate Retinitis pigmentosa registers worldwide are indispensable.
However; standardizations of the diagnosis, proper classification of the disease, and better therapeutic and rehabilitative approaches will help in better diagnosing of the disease, hence will help in bridging the gap between diagnosed and prevalent Retinitis pigmentosa population.
Downloads
Article in PDF
Recent Articles
- Actinium Announces SIERRA Trial Results; Santhera Seeks FDA Review for Vamorolone; Seres Announce...
- Cell and Gene Therapies in Rare Disorders: From Rarity to Recovery
- The Hidden World of Vision: Exploring the Top 5 Rare Eye Diseases
- Top 8 Breakthrough Gene Therapies for Retinitis Pigmentosa Treatment
- FDA Approves LUMAKRAS with VECTIBIX for KRAS G12C-Mutated Colorectal Cancer; PYC Receives FDA Rar...



