The Next Frontier in Bardet–Biedl Syndrome Treatment: Gene Therapy and Beyond
Feb 13, 2026
Table of Contents
Summary
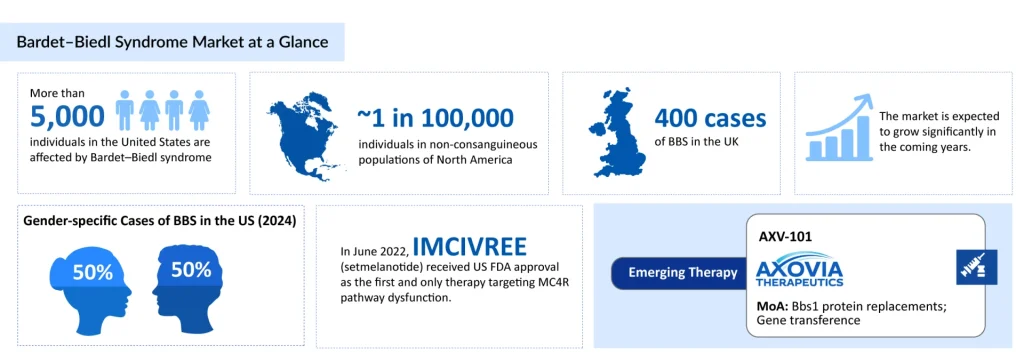
- Bardet–Biedl syndrome (BBS) is a rare, genetically heterogeneous disorder that affects more than 5,000 individuals in the United States.
- A major therapeutic milestone was the approval of IMCIVREE (setmelanotide), the first targeted MC4R agonist therapy addressing BBS-related obesity.
- Gene therapy with AXV-101, developed by Axovia Therapeutics, is emerging as a novel option for BBS1-related retinal degeneration.
Bardet–Biedl syndrome (BBS) is a rare, genetically heterogeneous disorder that affects more than 5,000 individuals in the United States. It occurs with equal frequency in males and females and has an estimated prevalence of approximately 1 in 100,000 in non-consanguineous North American populations. In Europe, the prevalence is estimated to range from approximately 1 in 125,000 to 1 in 160,000.
Downloads
Click Here To Get the Article in PDF

Recent Articles
As BBS manifests in so many ways, there is no single “cure.” Instead, treatment focuses on managing symptoms, preventing complications, and, more recently, targeting specific disease mechanisms such as obesity driven by melanocortin-4 receptor (MC4R) pathway dysfunction.
In recent years, the treatment landscape for BBS has evolved, with the first targeted therapy approved and gene therapy advancing into clinical trials.
IMCIVREE (Setmelanotide): The First Targeted Therapy for BBS
A major breakthrough in the BBS treatment landscape came with the approval of IMCIVREE (setmelanotide), a melanocortin-4 receptor agonist developed by Rhythm Pharmaceuticals (NASDAQ: RYTM). IMCIVREE targets the MC4R pathway, which is central to appetite regulation and energy expenditure. In BBS and other rare genetic obesity syndromes, dysfunction in this pathway contributes to early, severe obesity and pathological hyperphagia. Setmelanotide binds to and activates MC4R in the central nervous system, aiming to:
- Reduce excessive hunger and food-seeking behavior
- Promote weight loss and support long-term weight management
- Improve metabolic parameters tied to obesity
IMCIVREE initially received US FDA approval in 2022 as the first and only therapy targeting MC4R pathway dysfunction in BBS, indicated for chronic weight management in adults and children aged 6 years and older with BBS-related obesity.
In July 2024, the European Commission expanded marketing authorization to include children aged 2 to under 6 years with obesity associated with BBS. In December 2024, Rhythm Pharmaceuticals reported expanded regulatory approvals in both the United States and the United Kingdom, extending IMCIVREE’s use to children as young as 2 years of age for obesity and hunger control in BBS.
These expansions are significant because obesity and hyperphagia in BBS typically start very early in life. Intervening in children as young as 2 offers an opportunity to modify the trajectory of weight gain and metabolic complications.
IMCIVREE is a targeted, disease-specific therapy for the obesity component of BBS. However, it does not address other major manifestations such as:
- Retinal degeneration and vision loss
- Renal disease and structural anomalies
- Cognitive and behavioral challenges
Therefore, even with IMCIVREE, patients still require comprehensive supportive care. Nonetheless, for many families, having a therapy that directly reduces hunger and supports weight reduction represents a transformative change in day-to-day life and long-term health risk.
AXV-101 and Gene Therapy: A New Frontier for Vision in BBS
Beyond obesity, one of the highest unmet needs in Bardet–Biedl syndrome is the prevention of vision loss due to retinal dystrophy. DelveInsight analysts note that while metabolic complications dominate early disease management, retinal degeneration represents the most debilitating long-term morbidity, significantly impacting patient quality of life and independence. This is where gene therapy is emerging as a promising strategy. AXV-101, developed by Axovia Therapeutics, is an investigational gene therapy designed to treat blindness associated with BBS1 mutations. It aims to deliver a functional copy of the relevant gene to retinal cells, potentially preserving or restoring visual function.
AXV-101 has received US FDA Orphan Drug Designation (ODD) and Rare Pediatric Disease Designation (RPDD), reflecting the high unmet need and rarity of the condition. According to Ramandeep, Senior Consultant of Forecasting and Analytics at DelveInsight, the dual designation strategy positions Axovia to leverage substantial development incentives, including tax credits for clinical trials and potential pediatric priority review vouchers, while validating the translatable potential of their preclinical datasets. This Bardet–Biedl syndrome therapy is currently in early Phase I clinical development, with trial identifiers including CT.gov NCT07269665 and the UK registry ISRCTN96250868.
In May 2025, Axovia reported new preclinical data at the American Society of Gene & Cell Therapy (ASGCT) annual meeting, supporting continued progression into clinical studies. In January 2026, Axovia Therapeutics received an additional USD 1.1 million grant from Race Against Blindness, bringing the total funding to USD 3.1 million and enabling initiation of the AXIS (AXV-101) clinical trial in children with BBS type 1.
Ramandeep further concluded that this non-dilutive funding not only de-risks early clinical development but also demonstrates strong alignment with patient advocacy networks, a critical success factor for rare disease trials dependent on specialized recruitment and retention.
Although AXV-101 remains in early development and has not yet been approved, its progress underscores a broader shift: for the first time, BBS patients may benefit from disease-modifying ocular therapies rather than purely supportive measures.
Ramandeep views AXV-101’s entry into the clinic as part of a broader evolution in ocular gene therapy, leveraging lessons from approved retinal interventions like voretigene neparvovec while addressing the unique challenge of progressive ciliopathy-related degeneration. If clinical safety and preliminary efficacy signals are consistent across pediatric populations, AXV-101 could establish a critical precedent for genotype-specific BBS therapeutics and potentially expand the addressable market within the heterogeneous Bardet–Biedl syndrome landscape.
Unmet Needs in BBS Treatment
Despite these advances, major gaps remain in the BBS treatment landscape:
Single approved targeted therapy: IMCIVREE is currently the only approved disease-specific drug, and it addresses primarily obesity and hyperphagia. Retinal degeneration, renal decline, and other systemic complications still lack approved disease-modifying options.
Limited pipeline depth: Beyond AXV-101 and a small number of other exploratory programs, the BBS pipeline remains relatively thin relative to those for more common conditions. This is partly attributable to the syndrome’s rarity and genetic heterogeneity.
Delayed diagnosis and access to genetic testing: Many patients receive a definitive BBS diagnosis only after visual symptoms become apparent, thereby missing a critical window for early intervention. Expanding access to and awareness of genetic testing, along with the use of standardized diagnostic algorithms, is crucial for timely care. A case report of a 14-year-old boy with a classic BBS phenotype, including central obesity, post-axial polydactyly, and underdeveloped external genitalia, illustrates this critical gap, as he remained undiagnosed until the development of end-stage renal disease due to the unavailability of genetic testing in his region.
Long-term real-world data: As IMCIVREE and future BBS therapies are used over many years in children and adults, real-world evidence on efficacy, safety, adherence, and quality-of-life outcomes will be essential to refine treatment guidelines.
Continued collaboration between researchers, clinicians, patient advocacy groups, and industry innovators is essential to accelerate the development of new therapies targeting both obesity and non-obesity manifestations of BBS.
The Evolving Bardet–Biedl Syndrome Treatment Paradigm
Management of Bardet–Biedl syndrome has historically been dominated by supportive, organ-by-organ care delivered by a multidisciplinary team, with a focus on preserving vision, protecting renal function, managing obesity, and optimizing development. That foundation remains critical and will not go away.
The Bardet–Biedl syndrome treatment landscape is shifting with the emergence of mechanism-based therapies. MC4R agonism with IMCIVREE has introduced the first approved, targeted option for BBS-related obesity, now accessible to very young children across multiple major markets. At the same time, gene therapy approaches such as AXV-101 are opening the door to addressing the root cause of BBS-related blindness, signaling a new era of targeted intervention.
As these innovations advance and additional candidates enter the pipeline, the BBS treatment paradigm is likely to shift from purely supportive care toward a combination of symptom management plus disease-modifying interventions. For patients and caregivers, this holds the promise not just of better control of hunger and weight, but ultimately of preserved vision, improved organ function, and a significantly enhanced quality of life.
This optimism is grounded in recent preclinical findings showing that GLP-1 receptor agonists, already approved for diabetes and obesity, effectively target gut and brain pathways involved in feeding and metabolism, even in the context of BBS’s complex genetic disruption. In a Bbs5 knockout mouse model that closely mirrors human BBS symptoms, these therapies significantly improved metabolic function, reduced compulsive eating behavior, and normalized circulating hormone levels, despite the mice having impaired signaling from other key hormones such as insulin, leptin, and cholecystokinin.
Although clinicians currently show hesitancy to prescribe GLP-1 therapies to patients with BBS due to a lack of clinical trial data, these findings highlight the therapeutic potential for managing BBS-associated metabolic dysregulation and warrant further investigation for clinical application in both pediatric and adult populations.

Downloads
Article in PDF



