Huntington’s Disease Treatment Meets Its Match: Gene Therapy in Action
Nov 24, 2025
Table of Contents
Huntington’s Disease: Genetics, Symptoms, and Therapeutic Gaps
Huntington’s disease is often described as having Amyotrophic Lateral Sclerosis (ALS), Parkinson’s, and Alzheimer’s rolled into one—a neurodegenerative triple threat that devastates motor control, cognition, and behavior in a single blow. Caused by the expansion of CAG repeats in the HTT gene, the disease produces toxic huntingtin protein aggregates that progressively destroy neurons. Onset typically strikes between 30 and 50 years of age, though longer repeats can trigger the even more aggressive juvenile form (<20 years).
Inheritance is unforgiving: each child of an affected parent has a 50% risk, and paternal transmission often accelerates onset across generations through genetic anticipation. Once Huntington’s disease symptoms begin, chorea, psychiatric decline, and memory loss, the trajectory is inescapable. Diagnosis relies on clinical suspicion, confirmed by genetic testing and supported by counseling for families who must decide whether to undergo predictive testing.
Downloads
Click Here To Get the Article in PDF

Recent Articles
- Aktis’s Novel Targeted Alpha Radiopharmaceuticals; Koye Partners with Sonde Health; Novartis to S...
- Origins of the depressive behaviour in Huntington’s disease
- FDA Approves Sanofi/Regeneron’s DUPIXENT as First New CSU Therapy in Over a Decade; Gilead’s TROD...
- Bristol Myers Squibb’ Karuna Therapeutics Buyout; Orchard Therapeutics’ Lenmeldy FDA Approval; Ma...
- Notizia
Yet despite decades of research, Huntington’s disease remains incurable. Care today is multidisciplinary and supportive, encompassing physiotherapy, occupational therapy, and speech therapy, as well as psychiatric care, but the best it can offer is a reprieve. VMAT2 inhibitors such as AUSTEDO (deutetrabenazine) and INGREZZA (valbenazine) help regulate dopamine signaling and ease chorea, but they cannot slow neuronal death or alter the course of disease. Patients eventually become fully dependent on caregivers. The therapeutic gap is glaring: symptomatic relief without disease modification.
Global Impact: Expanding Huntington’s Disease Population and Clinical Demand
According to DelveInsight, around 82,000 individuals were living with Huntington’s disease across the major markets (the US, Germany, France, Italy, Spain, the UK, and Japan) in 2024. This number is expected to rise steadily, not because the disease is spreading, but because diagnosis is improving, supportive care extends survival, and awareness among clinicians and at-risk families is growing.
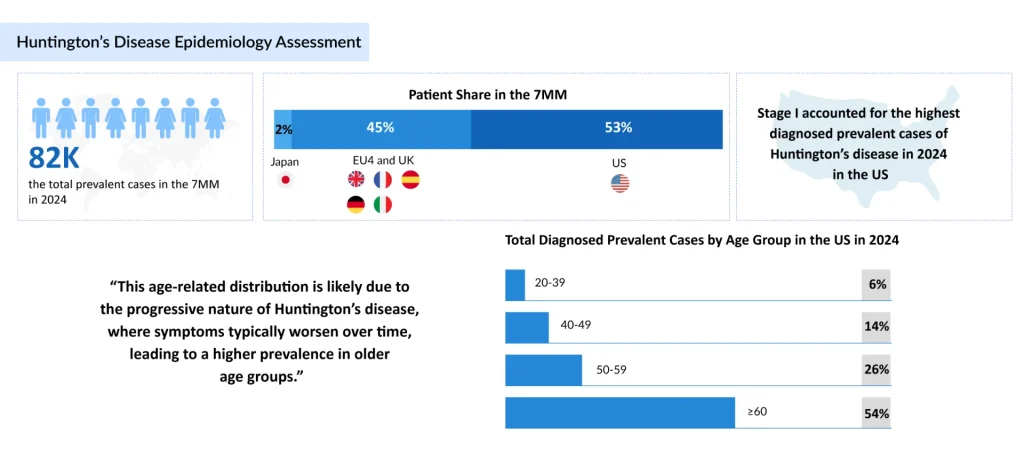
Aging populations are also unmasking late-onset Huntington’s disease cases, while genetic testing increasingly identifies pre-symptomatic carriers. Together, these forces ensure that prevalence and the demand for effective therapies will only continue to climb.
AMT-130: uniQure’s Bold Attempt to Rewrite Huntington’s Biology
For the first time, drug developers are aiming not at symptoms but at the genetic root. uniQure’s AMT-130 stands at the front of this revolution.
The strategy: use an AAV5 vector to deliver an artificial microRNA directly into the striatum—the brain’s epicenter of Huntington’s disease pathology. Administered via MRI-guided, convection-enhanced neurosurgery, the therapy silences both the full-length mutant HTT and the highly toxic exon-1 fragment. The vision is one-time dosing with long-lasting impact, resetting the disease trajectory at its source.
Regulatory Status
The US FDA has recognized AMT-130 for its potential to address the significant unmet medical needs in Huntington’s disease, receiving multiple expedited designations.
Breakthrough Therapy (April 2025): Acknowledges AMT-130’s potential to demonstrate substantial improvement over existing therapies.
Regenerative Medicine Advanced Therapy (RMAT, June 2024): The first RMAT designation for Huntington’s disease, facilitating closer collaboration with the US FDA and accelerated development.
Fast Track (April 2019): Supports faster review and more frequent interactions with the FDA to expedite development.
Orphan Drug (US and EU): Recognizes AMT-130 as a therapy for a rare condition, providing incentives for development and regulatory support.
Clinical Development Program
The AMT-130 program consists of multi-center, dose-escalating Phase I/II studies in the U.S. and Europe, enrolling patients with early manifest Huntington’s disease. Key aspects include
US study: 26 patients randomized to low dose (n = 6), high dose (n = 10), or sham procedure (n = 10). A blinded 12-month core study followed by a 5-year long-term follow-up.
European open-label study: 13 patients (low dose n = 6; high dose n = 7)
Additional cohorts evaluated dose escalation, combination with immunosuppression, and patients with lower striatal volumes.
Clinical outcomes were compared to a propensity score-matched external control derived from the Enroll-HD natural history study, acting as a virtual placebo to assess disease-modifying effects.
Topline Phase I/II Results
In September 2025, uniQure announced positive 36-month topline results for high-dose AMT-130.
| Measure/ Endpoint | AMT-130 High Dose Outcome | Expected Without Treatment (External Control) | Effect/Interpretation | Statistical Significance |
| Composite Unified Huntington’s Disease Rating Scale (cUHDRS) | –0.38 | –1.52 | 75% slower disease progression | p = 0.003 |
| Total Functional Capacity (TFC) | –0.36 | –0.88 | 60% slower decline in daily functioning | p = 0.033 |
| Symbol Digit Modality Test (SDMT) | -0.44 | –3.73 | 88% slower cognitive decline | p = 0.057 (trend) |
| Stroop Word Reading Test (SWRT) | 0.88 | –6.98 | 113% improvement / slower cognitive decline | p = 0.002 |
| Total Motor Score (TMS) | 2.01 | 4.88 | 59% slower worsening of motor symptoms | p = 0.174 (trend) |
| Cerebrospinal Fluid Neurofilament Light (CSF NfL) | –8.2% | – | Supports reduced neurodegeneration | – |
| Safety/Tolerability | Well-tolerated; procedure-related AEs resolved | – | Manageable safety profile | – |
Key Insights from the Results
- Significant slowing of disease progression across multiple functional, motor, and cognitive measures, particularly at high doses.
- Dose-dependent response observed: High dose consistently outperformed low dose.
- CSF NfL reduction supports the biological effect in slowing neurodegeneration.
- The safety profile was manageable, with adverse events largely associated with the administration procedure, all of which resolved.
Beyond uniQure: A Crowded and Creative Pipeline for Huntington’s Disease Treatment
While AMT-130 leads, it is far from alone. The Huntington’s disease clinical trial pipeline reflects a diverse, high-stakes race to crack the genetic code of neurodegeneration:
Roche/Spark Therapeutics – SPK-10001 (RG6662)
A microRNA therapy silencing HTT, now in Phase I/II, reflects Roche’s long-term bet following its USD 4.3 billion acquisition of Spark Therapeutics in 2019. Leveraging Spark’s gene therapy expertise, Roche is advancing SPK-10001, with the first patient already dosed in ongoing trials.
Latus Bio – LTS-201
Targets MSH3, a DNA repair enzyme that fuels somatic CAG expansion, rather than HTT directly. Preclinical data show reduced expansion in mice, and its AAV-DB-3 capsid has demonstrated efficient delivery to deep brain structures, long a bottleneck for gene therapy.
SOLA Biosciences – SOL-175
Uses proprietary JUMP70 technology to modulate the folding of mutant HTT’s polyQ stretches without disturbing normal HTT. Still preclinical, but it represents a precision strategy aimed at restoring protein balance.

VectorY Therapeutics – VTx-003
A vectorized antibody that selectively clears mutant HTT. In partnership with Shape Therapeutics, VectorY has access to novel AAV5-derived CNS capsids capable of deep-brain penetration via IV delivery—a potential game-changer for non-invasive administration.
Novartis/Voyager Therapeutics Collaboration
Through Voyager’s TRACER AAV platform, Novartis is evaluating next-generation capsids for CNS disorders, including an undisclosed Huntington’s disease program. The goal: overcome current distribution challenges and open the door to more efficient brain-wide delivery.
The Takeaway: From Symptom Control to Disease Modification
For decades, Huntington’s disease has epitomized therapeutic frustration: a devastating genetic disorder, relentless progression, and limited options beyond symptom management. That Huntington’s disease treatment landscape is now changing.
Gene therapies such as AMT-130 represent the first concerted efforts to alter the natural course of Huntington’s disease rather than merely mitigate its symptoms. The broader pipeline, including strategies to silence the HTT gene, block CAG expansions, or deploy vectorized antibodies, marks a new era of molecular neurology, focusing on addressing the root cause rather than its consequences.
The coming years will be critical in defining the regulatory and commercial path for Huntington’s disease gene therapies. For AMT-130, key near-term milestones include a pre-Biologics License Application (BLA) meeting with the US FDA in the fourth quarter of 2025, followed by a planned BLA submission in the first quarter of 2026 with a request for priority review. If approved, a US launch could take place later in 2026, potentially marking the first disease-modifying therapy for Huntington’s disease.
Should these programs fulfill their promise, the field could finally shift from merely “managing decline” to changing the disease trajectory—a transformative moment for patients, families, and the future of neurodegenerative medicine.

Downloads
Article in PDF
Recent Articles
- AbbVie Reveals Phase III TEMPO-2 Trial Positive Topline Results; FDA Accepts GSK’s NUCALA Submiss...
- Origins of the depressive behaviour in Huntington’s disease
- Aktis’s Novel Targeted Alpha Radiopharmaceuticals; Koye Partners with Sonde Health; Novartis to S...
- Bristol Myers Squibb’ Karuna Therapeutics Buyout; Orchard Therapeutics’ Lenmeldy FDA Approval; Ma...
- FDA Approves Sanofi/Regeneron’s DUPIXENT as First New CSU Therapy in Over a Decade; Gilead’s TROD...



