A Turning Point: INGREZZA’s Impact on Huntington’s Disease Treatment and the Rise of Novel Therapies
Sep 18, 2023
Table of Contents
Huntington’s disease is an incurable, rare genetic, progressive neurodegenerative disorder. According to the National Organization for Rare Disorders (NORD), about 30,000 people in the United States have Huntington’s disease, and another 200,000 are at risk of developing the condition.
As per DelveInsight analysis, In the year 2022, the total prevalent cases of Huntington’s disease were found to be ~81,000 in the 7MM. As per the estimates, in 2022, the total prevalent cases of Huntington’s disease were 43,000 in the US. These cases are expected to grow within the study period (2019–2032).
Downloads
Click Here To Get the Article in PDF

Recent Articles
- Aktis’s Novel Targeted Alpha Radiopharmaceuticals; Koye Partners with Sonde Health; Novartis to S...
- Huntington’s Disease Treatment Meets Its Match: Gene Therapy in Action
- SpringWorks’s Desmoid Tumors Therapeutic, Nirogacestat; Orphan Drug Designation to AskBio’s AB-10...
- KINECT 4 Study; KindredBio Announces Results; Mylan and Aspen’s Launch; EMA investigates; Amgen’s...
- Bristol Myers Squibb’ Karuna Therapeutics Buyout; Orchard Therapeutics’ Lenmeldy FDA Approval; Ma...
So far, only two drugs have been approved by the FDA for Huntington’s disease treatment, but they do not treat the disease, instead, they are used to treat symptoms like chorea associated with Huntington’s disease. The first was tetrabenazine, which was approved by the US FDA in 2008 based on the TETRA study. This drug is sold as XENAZINE by Lundbeck. The second was AUSTDEO (deutetrabenazine; Deuterated version of tetrabenazine), which was approved by the US FDA in 2017. Both medicines have been approved to treat Huntington’s disease chorea. XENAZINE was the first drug ever approved in the US for the treatment of Huntington’s disease symptoms. This medication was approved with a premium price tag in the US due to its Orphan Drug designation. XENAZINE has significant side effects such as somnolence, insomnia, Parkinsonism, severe depression, fatigue, anxiety, and irritability.
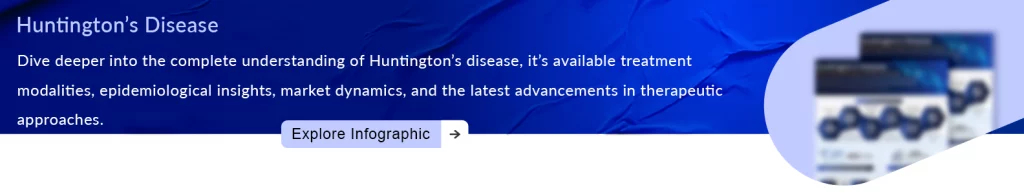
According to DelveInsight’s analysts, tetrabenazine has notable flaws that Teva’s AUSTEDO could potentially address. While Austedo has been designated a new molecular entity and an orphan drug, it was approved through the 505(b) pathway with tetrabenazine as the Reference Listed Drug (RLD). In February 2023, the US FDA-approved extended-release deutetrabenazine (AUSTEDO XR) tablets for adult patients with tardive dyskinesia and chorea associated with Huntington’s disease.
Approval of INGREZZA for Huntington’s Disease Treatment
Ingrezza from Neurocrine Biosciences is poised to compete with Teva’s Austedo, following its recent FDA approval. This opens the door to a treatment area with the potential for hundreds of millions of dollars in revenue. The FDA’s green light allows Ingrezza capsules to be used in the treatment of chorea associated with Huntington’s disease in adults. Ingrezza initially gained approval for tardive dyskinesia treatment in 2017. With this second approval, Ingrezza’s label now closely aligns with that of Teva Pharmaceutical Industries’ Austedo, which also received approval for Huntington’s disease chorea in 2017.
INGREZZA stands out as the sole selective vesicular monoamine transporter 2 (VMAT2) inhibitor that allows for flexible dosing based on a patient’s response and tolerance, without the need for complex titration. With INGREZZA, it’s always a straightforward one-capsule, once-daily regimen. Its FDA approval is backed by extensive clinical research, including studies conducted in collaboration with the Huntington Study Group (HSG), such as the KINECT®-HD Phase III trial and the ongoing KINECT®-HD2 open-label extension study.
“We are delighted to introduce INGREZZA to individuals affected by Huntington’s Disease, as well as their devoted caregivers. This groundbreaking, once-daily, single-capsule treatment has exhibited remarkable efficacy in alleviating HD chorea during clinical trials,” expressed Kevin C. Gorman, CEO of Neurocrine Biosciences. “We extend our heartfelt gratitude to the Huntington’s Disease community for their invaluable contributions to this significant achievement, and our dedication to providing solutions for individuals with unmet medical needs in the realm of neurological disorders remains unwavering.”
In the late-phase KINECT-HD trial, Ingrezza met its primary endpoint by demonstrating a reduction in the Unified Huntington’s Disease Rating Scale total maximal chorea score compared to baseline during Weeks 10 and 12. Patients receiving Ingrezza experienced an average improvement of 3.2 points on the 28-point scale, marking a significant reduction in chorea severity when contrasted with the placebo group. Noteworthy is Ingrezza’s achievement of blockbuster status, as it generated $1.43 billion in sales for Neurocrine last year. In February, Neurocrine initially anticipated Ingrezza’s sales to range between $1.67 billion and $1.77 billion in 2023. However, the company later adjusted this estimate to a range of $1.77 billion to $1.82 billion in August.
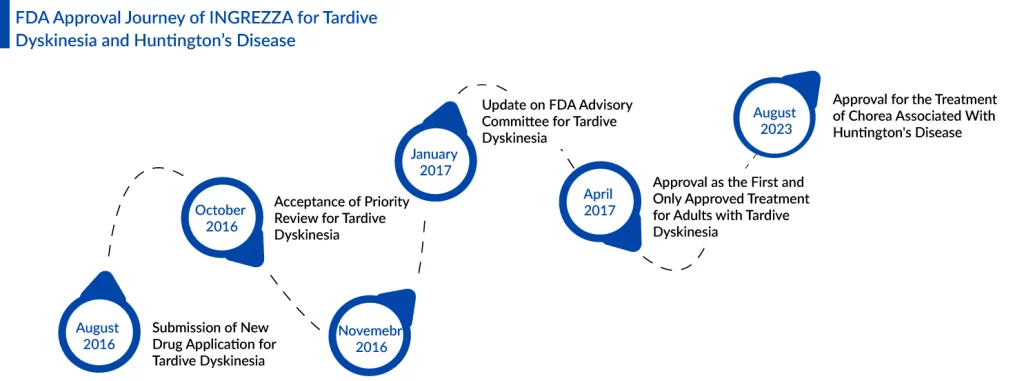
Promising Therapies in Pipeline for Huntington’s Disease Treatment
Key player such as Prilenia Therapeutics [Pridopidine (ACR-16; Huntexil)] is involved in the development of Phase III Huntington’s disease drugs. Whereas, Phase II drugs are being developed by companies like Annexon Biosciences [ANX005], SOM Biotech [SOM3355/Bevantolol], Vaccinex [Pepinemab (VX15/2503)], and others. Apart from these, other companies are also investigating their candidates to manage Huntington’s disease in the 7MM.
Pridopidine (formerly Huntexil) is Prilenia Therapeutics’ lead asset and a first-in-class selective and potent Sigma-1-receptor (S1R) agonist with neuroprotective properties. Activation of the S1R by pridopidine enhances the clearance of toxic proteins, enhances energy production, and reduces cellular stress and inflammation. Prilenia has also received the Fast Track Designation for Pridopidine for the treatment of Huntington’s Disease. Currently, Pridopidine is being evaluated in the ongoing Phase III (NCT04556656) global clinical trial PROOF-HD (PRidopidine Outcome on Function in Huntington’s disease) in collaboration with the Huntington Study Group (HSG). As per the company, the successful outcome of PROOF-HD may lead to Pridopidine’s approval as a treatment for Huntington’s disease.
Som Biotech’s Phase II candidate SOM3355 (bevantolol) is another VMAT2 inhibitor being investigated for chorea associated with Huntington’s disease. According to the company, SOM3355 differs from Tetrabenazine in interaction with VMAT2 and inhibition of VMAT1, resulting in a balanced shift in monoamine signaling that alleviates motor symptoms of Huntington’s disease while maintaining mental well-being. In October 2021, SOM Biotech announced that the FDA had granted Orphan Drug Designation (ODD) for SOM3355 for the treatment of chorea movements in Huntington’s disease. A Phase II (NCT03575676) trial for this drug in Huntington’s disease chorea has been completed.
ANX005 is a clinical-stage investigational monoclonal antibody intended to treat patients with antibody-mediated autoimmune and complement-mediated neurodegenerative disorders. This novel therapy is formulated for IV administration and is designed to inhibit C1q and the entire classical complement pathway. A Phase II (NCT04514367) trial of this drug for Huntington’s disease has been completed. In October 2021, Annexon was granted orphan drug designation for the treatment of Huntington’s disease by the FDA.
VX15/2503 (pepinemab) is a novel humanized monoclonal IgG4 antibody that binds and blocks the signaling activity of SEMA4D. SEMA4D is an extracellular signaling molecule that regulates the migration and activation of immune and inflammatory cells at sites of injury and cancer. Preclinical testing has demonstrated the beneficial effects of anti-SEMA4D treatment in a variety of neurodegenerative disease models. Vaccinex is committed to the development of this important antibody that has the potential to help people with different neurodegenerative disorders that share common mechanisms of pathology. Vaccinex has completed the Phase II (NCT02481674; SIGNAL-HD) clinical trial to evaluate the effect of pepinemab in both early manifest and late prodromal patients with Huntington’s disease. The endpoints of the study included safety and tolerability, clinical and behavioral outcomes, as well as measures of brain volume, metabolic activity, and inflammation. In August 2016, the FDA granted Fast-track Designation and Orphan drug designation to pepinemab for Huntington’s disease treatment.
Unlocking the Potential of Gene Therapy for Huntington’s Disease Treatment
Gene therapy for Huntington’s disease treatment is at the forefront of groundbreaking medical research. By addressing the genetic root cause of this devastating neurodegenerative disorder, gene therapy offers a glimmer of hope for patients and their families. Huntington’s disease, an inherited condition with no known cure, is characterized by relentless deterioration of physical and cognitive abilities. Currently, only UniQure Biopharma is working in the Huntington’s disease treatment space with its lead asset AMT-130.
AMT-130 consists of an AAV5—vector carrying an artificial microRNA specifically tailored to silence the huntingtin gene, leveraging the proprietary miQURE silencing technology. The therapeutic goal is to inhibit the production of the mutant protein (mHTT). Using AAV vectors to deliver microRNAs (miRNAs) directly to the brain for non-selective knockdown of the huntingtin gene represents a highly innovative and promising approach to treating Huntington’s disease. The drug has received orphan drug designation and fast-track designation from the FDA for Huntington’s disease treatment and is currently in Phase I/II (NCT05243017, NCT04120493) study of clinical development. The drug has been granted orphan drug designation by the FDA and the EMA and has also been granted the fast track designation by the FDA for Huntington’s disease treatment.
Expected Roadblocks and Future of Huntington’s Disease Treatment
The lack of approved therapy for Huntington’s disease treatment is not just a limitation for the patients but for the pharmaceutical giants as well. Undoubtedly, companies are trying to do their part by developing much-needed therapies, however, the high failure rate of emerging therapies for Huntington’s disease treatment is the major barrier. Not just one or two, but several candidates have failed so far. Both the developer and Huntington’s community was optimistic about a few candidate which eventually turned out to be the opposite. The biggest blow came with the failure of late-stage candidate tominersen.
Recently, Wave Life Sciences announced that their two antisense oligonucleotides (ASOs) in Phase I/II studies in Huntington’s disease patients did not successfully reduce mutant huntingtin levels, hence the company decided to discontinue those products. Roche/Ionis antisense asset tominersen had the same fate. The failure of Roche and Ionis’s late-stage candidate tominersen had already cast doubt on Wave’s approach. Development and approval of a novel therapy that treat Huntington’s disease is a major unmet need, and at present, a handful of companies are working toward that goal.
Moreover, recent advancements in gene therapy are changing the landscape of Huntington’s disease treatment. Innovative approaches, including gene silencing, gene editing, and gene replacement, are being developed to combat the aberrant HTT gene responsible for the production of the toxic mutant huntingtin protein. These therapies aim not only to halt the progression of the disease but also to potentially reverse its devastating effects. While challenges such as efficient brain delivery, long-term efficacy, safety concerns, and ethical considerations remain, the relentless pursuit of solutions is reshaping the future of Huntington’s disease treatment. Collaborative efforts between researchers, healthcare professionals, and Huntington’s disease community provide the foundation for unlocking the potential of gene therapy, holding the promise of a brighter future for those affected by this challenging condition.

Downloads
Article in PDF
Recent Articles
- KINECT 4 Study; KindredBio Announces Results; Mylan and Aspen’s Launch; EMA investigates; Amgen’s...
- Origins of the depressive behaviour in Huntington’s disease
- Notizia
- Huntington’s Disease Treatment Meets Its Match: Gene Therapy in Action
- Aktis’s Novel Targeted Alpha Radiopharmaceuticals; Koye Partners with Sonde Health; Novartis to S...



