UCB’s KYGEVVI Approval: A Breakthrough for Thymidine Kinase 2 Deficiency
Nov 10, 2025
In a significant milestone for the rare disease community, the FDA approved KYGEVVI (doxecitine and doxribtimine) in November 2025, marking the first and only approved treatment for thymidine kinase 2 deficiency (TK2d). This breakthrough approval represents a transformative moment for patients and families facing this ultra-rare, life-threatening genetic mitochondrial disease that previously had no treatment options beyond supportive care.
KYGEVVI has been approved for use in both adults and pediatric patients who develop symptoms by the age of 12. TK2d is an exceptionally rare genetic mitochondrial disorder affecting fewer than 2 people per 1,000,000 individuals worldwide. The disease is caused by mutations in the TK2 gene, which normally produces a protein essential for maintaining proper mitochondrial function. In patients with TK2d, this critical organelle deteriorates over time, becoming unable to generate sufficient energy for cells throughout the body.
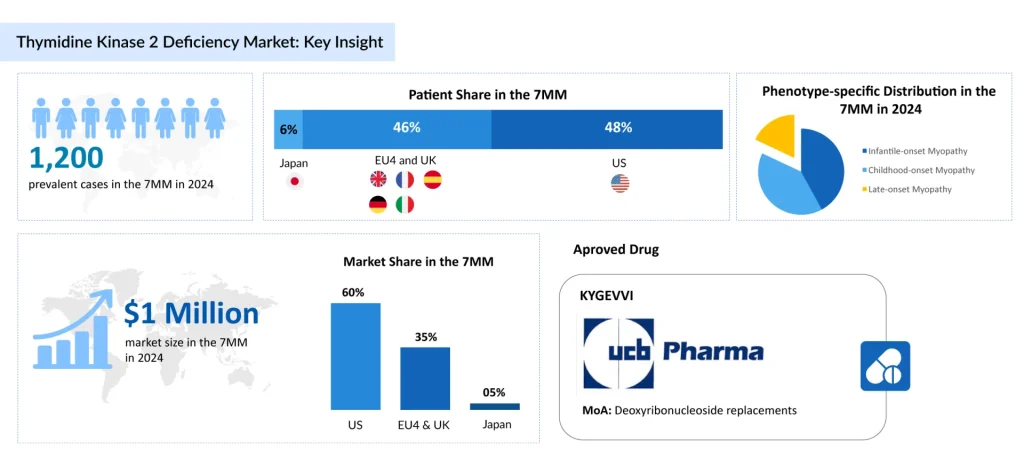
Early-onset TK2d is associated with a higher risk of rapid disease progression and mortality, often proving fatal within three years of symptom onset. DelveInsight’s analysis estimates that there were around 1,200 prevalent cases of TK2d across the seven major markets in 2024. This figure is expected to rise over the 2025–2034 forecast period, fueled by advances in genetic testing and broader healthcare access.
Downloads
Click Here To Get the Article in PDF

KYGEVVI is an oral formulation containing the pyrimidine nucleosides doxecitine and doxribtimine, which incorporate into mitochondrial DNA within skeletal muscle. This strategy has demonstrated the ability to restore mitochondrial DNA copy numbers in TK2d mouse models.
The approval of KYGEVVI represents far more than a regulatory achievement—it signals hope and tangible medical progress for a severely affected patient community. According to Kristen Clifford, president and CEO of the United Mitochondrial Disease Foundation, “It’s hard to overstate the importance of this FDA approval for those diagnosed with TK2d. This is an ultra-rare disease community in dire need of treatment options. For too long, caregivers and their families have had to endure the burden of this disease. Having the first-ever FDA-approved therapy for TK2d in this patient population not only meets a critical medical need – it represents something greater – hope for the future.”
Dr. Michio Hirano, a leading mitochondrial disease researcher and Chief of the Division of Neuromuscular Medicine at Columbia University Irving Medical Center, echoed this sentiment, stating, “I’ve been studying mitochondrial diseases for more than three decades and have witnessed firsthand the impact TK2d has on patients and their families. We have been waiting for an approved treatment for many years, and this approval marks a significant milestone in how we can support and manage this debilitating condition.”
The FDA approval of KYGEVVI is based on safety and efficacy data from a Phase II TK2d clinical trial, two retrospective chart reviews, and an expanded access program. Altogether, these studies evaluated 82 unique patients treated with KYGEVVI or pyrimidine nucleosides who had TK2d symptom onset at or before 12 years of age. Efficacy was determined by comparing overall survival between treated pediatric and adult patients and an external control group of untreated patients matched by age at symptom onset (≤2 years or >2 to ≤12 years).
In total, 78 matched pairs were analyzed, revealing that KYGEVVI treatment reduced the risk of death by approximately 86% (95% CI: 61%, 96%) and significantly improved survival time from treatment initiation. Among the treated patients, the median age of symptom onset was 1.5 years (range: 0.01–12 years), with a median treatment duration of 4 years (range: 1 day–12 years) and a median daily dose of 762 mg/kg (range: 260–800 mg/kg). The most frequent adverse reactions (≥5% incidence) included diarrhea, abdominal pain (including upper abdominal pain), vomiting, and elevated liver enzymes (ALT and AST).
A regulatory review of doxecitine and doxribtimine is currently in progress at the European Medicines Agency (EMA), with additional global submissions planned. KYGEVVI is not yet approved outside the United States, but UCB anticipates US commercial availability in Q1 2026. To ensure equitable access, UCB will introduce a personalized patient support program designed to prioritize the needs of patients and caregivers.
In the US, KYGEVVI has been granted Orphan Drug, Breakthrough Therapy, Priority Review, and Rare Pediatric Disease designations by the FDA. With this approval, UCB has also received a Rare Pediatric Disease Priority Review Voucher (RPDPRV), which can be used to obtain priority review for a future marketing application.
UCB has experienced strong growth driven by BIMZELX, approved for plaque psoriasis in 2023, which brought in €799 million ($918 million) in sales during the first half of the year. The company’s performance was further boosted by its 2023-approved myasthenia gravis treatments, RYSTIGGO and ZILBRYSQ, which together generated €239 million ($274 million) over the same period. Overall, UCB reported a 25% revenue increase in the first half of the year and achieved the largest market capitalization gain, 41%, among the industry’s top 20 companies in the third quarter.
In conclusion, the FDA approval of KYGEVVI represents a watershed moment in the treatment of TK2d and exemplifies the progress being made in ultra-rare disease therapeutics. By providing an 86% reduction in mortality risk for patients with this previously untreatable condition, KYGEVVI offers clinically meaningful benefits that can extend and improve the quality of life for affected individuals and their families.
As the pharmaceutical industry continues to focus on addressing unmet needs in rare disease populations, KYGEVVI serves as a compelling example of how targeted drug development, rigorous clinical evidence, and expedited regulatory pathways can transform the landscape for patients facing even the rarest of conditions.




