Leigh Syndrome Treatment: The Ongoing Quest for an Effective Cure
May 26, 2025
Table of Contents
Leigh syndrome, also called subacute necrotizing encephalomyelopathy, is a rare and serious genetic neurometabolic condition classified as a primary mitochondrial disease. It is associated with mutations in more than 75 genes, most of which are involved in cellular energy production. However, because many cases do not have a confirmed genetic cause, the list of implicated genes is likely incomplete. Although some acquired disorders may resemble Leigh syndrome, a diagnosis requires definitive evidence of an underlying mitochondrial dysfunction.
The condition affects roughly 1 in 40,000 people. The classic form of Leigh syndrome represents about 80% of diagnosed cases. Among nuclear DNA mutations, the SURF1 gene is the most commonly involved, accounting for 72% of those cases, with mutations identified in a total of 18 different nuclear genes. In mitochondrial DNA, MT-ATP6 mutations are the most prevalent, found in 55% of cases, with six other mitochondrial genes also implicated.
Downloads
Click Here To Get the Article in PDF

Leigh Syndrome Treatment: A Focus on Care, Not Cure
Leigh syndrome currently lacks an approved cure, and Leigh syndrome treatment is mainly supportive, focusing on individualized care to relieve symptoms, slow disease progression, and enhance quality of life. A central aspect of management involves controlling lactic acidosis, a hallmark of the disorder that contributes to fatigue, muscle weakness, and respiratory issues. Nutritional support is often necessary, with gastrostomy (G-tube) placement commonly used in children who have feeding difficulties or elevated caloric needs due to metabolic demands.
While most patients receive symptom-based care, certain individuals with specific genetic mutations may benefit from targeted Leigh syndrome therapies. For instance, those with mutations in the SLC19A3 gene, linked to biotin-thiamine-responsive basal ganglia disease, often respond to high doses of thiamine and biotin. Likewise, patients with BTD gene mutations causing biotinidase deficiency can improve with biotin supplementation. In cases involving pyruvate dehydrogenase complex deficiency, a ketogenic diet, high in fat and low in carbohydrates, along with thiamine, has shown therapeutic promise.
Other commonly used interventions include vitamins and cofactors such as thiamine (vitamin B1), coenzyme Q10, L-carnitine, and multivitamin combinations, although clinical evidence for their effectiveness is limited. Antiepileptic medications are frequently used to manage seizures, and supportive treatments may also involve vision care, respiratory support, and therapies for spasticity and involuntary movements like dystonia.
Due to the diverse genetic origins of Leigh syndrome, stemming from mutations in either nuclear or mitochondrial DNA, genetic testing and counseling are vital for informed family planning and long-term management. Despite current care options, the overall prognosis remains poor, especially in early-onset cases. Data from retrospective studies show high mortality rates in childhood, with median survival for neonatal-onset mitochondrial forms being as short as 90 days. These findings highlight the urgent need for the development of disease-modifying Leigh syndrome treatments.
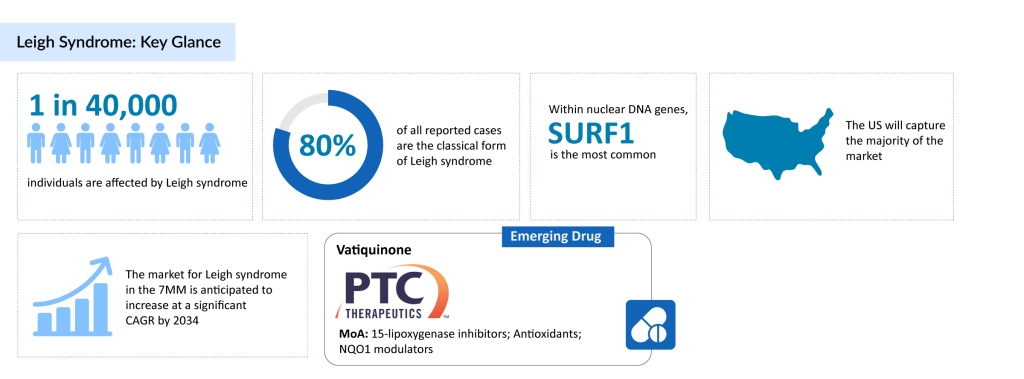
PTC Therapeutics Takes the Lead in Leigh Syndrome Drug Development
The treatment pipeline for Leigh syndrome remains sparse, with only a handful of companies pursuing therapeutic options. One notable contender is PTC Therapeutics, which is developing Vatiquinone as a potential Leigh syndrome treatment.
Vatiquinone (PTC743) is an investigational drug that inhibits 15-lipoxygenase, an enzyme involved in regulating key energy production and oxidative stress pathways. It has been tested in several clinical trials, showing potential in reducing mortality risk and alleviating various neurological and neuromuscular symptoms.
However, in June 2023, the Phase II/III MIT-E trial evaluating Vatiquinone for mitochondrial disease-associated seizures (MDAS) did not meet its primary goal of reducing observable motor seizures. Despite this setback, Vatiquinone continues to be studied in a Phase III trial targeting inherited mitochondrial diseases, including Leigh syndrome.
Bridging the Gaps in Leigh Syndrome Care
Leigh syndrome represents a significant area of unmet medical need, driven by its rapid neurodegenerative progression, complex genetic basis, and the absence of curative treatments. Most patients experience early-onset symptoms such as psychomotor decline, seizures, and respiratory complications, often leading to poor survival outcomes. Current Leigh syndrome therapies are primarily supportive, while investigational approaches like gene therapy and NAD+ supplementation remain in early development. The disorder’s genetic and clinical diversity complicates both diagnosis and the delivery of personalized care. Moreover, its multisystem nature demands a coordinated, multidisciplinary approach that is often lacking. There is an urgent need for effective Leigh syndrome treatments, improved diagnostic tools, and integrated care strategies to ease the burden on affected individuals and their families.
Additionally, drug development for Leigh syndrome treatment is particularly challenging due to its extensive genetic and biochemical variability, rooted in mitochondrial dysfunction. This heterogeneity makes it difficult to pinpoint universal therapeutic targets and to design trials that yield meaningful results. The rarity of the condition and the globally scattered patient population also pose significant hurdles for clinical trial enrollment. Furthermore, progress is hindered by a limited number of relevant preclinical models and an incomplete understanding of the disease’s mechanisms. While some promising candidates like vatiquinone have progressed to advanced clinical stages, the overall Leigh syndrome drug development pipeline remains thin, underscoring the substantial scientific and logistical obstacles in this field.
Beyond the Horizon: What Lies Ahead for Leigh Syndrome Treatments?
Leigh syndrome, a rare and often fatal neurodegenerative disorder, has historically posed significant challenges for clinicians and researchers due to its genetic complexity and early onset. Caused by mutations in mitochondrial or nuclear DNA affecting energy production, the syndrome currently lacks curative therapies, with treatment largely focused on managing symptoms and slowing disease progression.
However, recent advances in molecular genetics and targeted therapeutics are beginning to shift the landscape. Mitochondrial replacement therapy, gene therapy, and precision medicine approaches offer new hope. Efforts like the development of adeno-associated virus (AAV) vectors for gene delivery and the use of small molecule therapies to enhance mitochondrial function are already in preclinical or early clinical stages, signaling a turning point in the field.
Looking ahead, the future of Leigh syndrome treatment lies in continued investment in translational research, improved diagnostic tools, and global collaboration among scientists, clinicians, and patient advocacy groups. Personalized treatment strategies tailored to the specific genetic mutations of patients could dramatically improve outcomes. Additionally, initiatives to better understand disease mechanisms through stem cell models and AI-driven drug discovery are likely to accelerate the identification of novel targets.
While challenges remain, the growing pipeline of innovative therapies marks a promising new chapter in the fight against Leigh syndrome, bringing a future of possibility to patients and families long underserved by conventional medicine.

Downloads
Article in PDF



