Netherton Syndrome: Unraveling the Uncertainties of a Rare Genetic Skin Disorder
Sep 29, 2023
Table of Contents
Quick Facts about Netherton Syndrome
- Netherton syndrome, a less common form of ichthyosis, is a monogenic cutaneous condition characterized by congenital scaly erythroderma, evolving into typical erythematous patches with peripheral scaling, hair shaft abnormalities, and atopic manifestation.
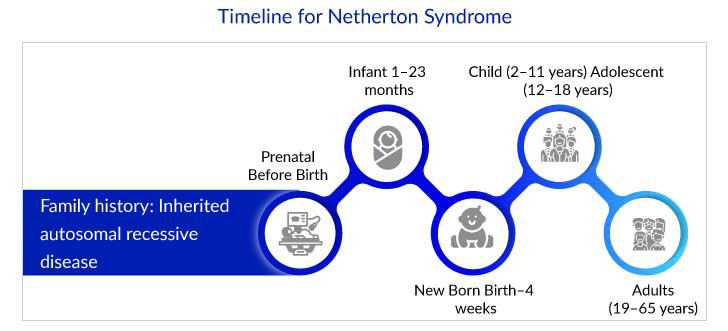
- Netherton syndrome can appear at birth or within the first few weeks of life.
- It is caused by deleterious mutations in the SPINK5 gene encoding the LEKTI1, resulting in elevated cytokine levels in the IL-17/IL-36 pathway.
- The existing pharmacological therapies for Netherton syndrome are symptomatic and include emollients, keratolytics, and oral retinoids. The efficacy of these treatments are moderate and usually ineffective against skin inflammation.
- Netherton syndrome significantly decreases the QoL of affected individuals and their families, resulting in emotional stress and social stigma caused due to visible skin manifestations.
- In the coming years, gene therapies and other novel therapies are expected to be developed to fulfill the unmet treatment needs for Netherton syndrome.
Netherton syndrome is a rare autosomal recessive illness characterized by congenital ichthyosiform erythroderma (CIE) or ichthyosis linearis circumflexa (ILC), hair shaft abnormalities, and atopic diathesis (elevated serum IgE). The syndrome is caused by mutations in the serine peptidase inhibitor Kazal type 5 (SPINK5) gene, which plays a crucial role in skin barrier function and immune regulation. Further SPINK5 gene encodes the protein serine peptidase inhibitor lymphoepithelial Kazal-type-related inhibitor (LEKTI). Loss of LEKTI causes epidermal protease dysregulation and significant skin barrier deterioration. Kallikrein-related peptidases (KLK), such as KLK5, KLK7, KLK14, and the epidermal elastase 2 (ELA2), which the LEKTI suppresses, have been linked to Netherton syndrome pathophysiology.
Only a handful of patients get diagnosed with Netherton syndrome, mostly based on clinical symptoms, family history, and genetic testing that identify mutations in the SPINK5 gene. Other types of testing, such as a careful study of the hair and a skin biopsy are also useful in determining the cause. Visibility of red skin at birth can indicate various illnesses, such as other types of ichthyosis, severe eczema, or other forms of immune deficiency; newborns and children are frequently not identified for months or years. Skin biopsy and DNA testing are also performed to confirm the diagnosis. Thus, a differential diagnosis plays a critical role.
Downloads
Click Here To Get the Article in PDF

Understanding SPINK5 Mutations and LEKTI Deficiency
The skin acts as a protective barrier, preventing water loss and allergens, irritants, and infections from entering the body. A disruption of the epidermal barrier plays an important role in developing several major skin diseases, including ichthyosis, psoriasis, atopic dermatitis, and Netherton syndrome. The underlying pathomechanism of these disorders is influenced by disruptions in the epidermal barrier functioning and immunological alterations of the skin.
SPINK5 gene is necessary to maintain skin barrier integrity. LEKTI deficiency results in defective barrier functioning as LEKTI inhibits specific enzymes (serine proteinases) in the skin’s outermost layer (epidermis). The serine proteinases break down the intracellular cement that holds together the stratum corneum cells in the epidermis, allowing the skin to shed cells (a process known as cell desquamation). LEKTI deficiency causes unrestrained desquamation of the stratum corneum cells, resulting in red and scaly skin. The skin’s barrier function is compromised along with a rise in permeability while its capacity to bind water declines, further leading to dryness.
As per DelveInsight estimates, the total diagnosed prevalent cases of Netherton syndrome in the US were around 1,700 in 2022. The life expectancy of infants with Netherton syndrome is low due to complications associated with the disease. Additionally, they may suffer from a high risk of life-threatening problems such as hypernatremic dehydration, failure to thrive, and sepsis in the first and second years of life. However, the health of most children will start to improve, but they remain underweight and short-stature.
Unmet Needs: A Call for Netherton Syndrome Treatment Improvement
As Netherton syndrome is a rare genodermatosis, it has an inherent disadvantage in its drug development. The lack of understanding about Netherton syndrome is a major issue affecting patients, families, healthcare professionals, and society. Several causes contribute to this lack of awareness, including overlapping symptoms during early infancy with other skin illnesses and underdiagnosis or misinterpretation.
“Netherton syndrome is frequently confused with atopic dermatitis. Dermoscopy can help narrow the diagnosis by detecting trichorrhexis invaginata or hair banding. The exact molecular therapy of Netherton syndrome with kallikrein 5 inhibitors is being researched.”
MD, American Academy of Dermatology, US
The Netherton syndrome therapy landscape currently lacks any FDA-approved therapies, diagnostic, or treatment guidelines. Although off-label medicines for Netherton syndrome treatment are available, they are constrained by several obstacles and safety concerns that do not address the underlying cause of the disease and are focused on symptom management, such as reducing dryness, itching skin, outbreaks of red, scaly, circular rashes, and controlling inflammation.
The current Netherton syndrome treatment regime includes bathing often with a mild, soap-free cleaner to soften the skin, which may benefit Netherton syndrome patients. Regular use of emollients and moisturizing creams containing petrolatum or lanolin and/or skin barrier repair therapies, including ceramides or cholesterol, for patient care. Topical creams and steroids, such as topical calcineurin inhibitors (pimecrolimus and tacrolimus), may be beneficial, but they must be used with caution because part of the drugs is absorbed into the circulation and induce itch and discomfort that is then relieved with the use of steroids. This should not be used in large doses. Because the skin is so thin, treatments to exfoliate or remove scale, such as alpha-hydroxy acids (lactic acid, glycolic acid), salicylic acid, and oral retinoids, are ineffective and may aggravate the symptoms. Thus, patients suffering from rare skin diseases have few or no treatment options. Furthermore, an early diagnosis is essential for proper patient management. Clinical characteristics, family history, and genetic testing to find SPINK5 gene mutations are used to diagnose Netherton syndrome. It may also be helpful to do further tests, such as a thorough hair examination or a skin biopsy, to acquire a small skin sample for microscopic analysis. Even though genetic testing is reliable, it is expensive and not always widely accessible in hospitals. A skin biopsy can be painful for pediatric patients and is time-consuming.
Emerging Therapies and Research Frontiers in Netherton Syndrome Treatment
Over the past few years, Netherton syndrome treatment options have not evolved much. The current treatment landscape lacks licensed therapies and treatment guidelines; however, off-label therapies such as emollients, moisturizers, topical corticosteroids, calcineurin inhibitors, and oral retinoids (acitretin) or immune-modulating medications are used to reduce inflammation and itching. Also, their long-term use is associated with various challenges and safety concerns. Furthermore, effective symptom management necessitates a multidisciplinary strategy that focuses on the distinctive clinical characteristics of individual patients.
Though the pipeline for Netherton syndrome treatment is limited, small molecules and monoclonal antibodies (mAb) are major molecule types currently in trials. The small molecules bind to specific sections of the targeted protein and thus block KLK activation caused due to malfunctioning of the SPINK5 gene in Netherton syndrome patients. Biological therapy includes the use of monoclonal antibodies that target specific markers, such as tumor necrosis factor-alpha (TNF-α), Intravenous immunoglobulins (IVIG), interleukin-13 (IL-13), interleukin-17 (IL-17), and interleukin-36 (IL-36). They may be beneficial in treating Netherton syndrome and several ichthyosis subtypes; however, the response is mostly seen as reducing the inflammatory component and pruritus. Further, the effect on scaling seems more variable and has given new ground to repurpose biologicals that can further help predict which phenotypically and genetically subtypes show a better response to anti-IL-17 therapy. Additionally, the use of repurposed biologics such as IV immunoglobulins and anti-IL-17A blockers in pediatric Netherton syndrome patients have demonstrated the efficacy of various biotherapies targeting IL-17A, IL-12/IL-23, IL-4R, and IL-13R, TNF-α, and IL-1. For example, SPEVIGO (spesolimab/BI 655130) is an IL-36R antagonist that acts upon various types of cells, including keratinocytes, epithelial cells, and immune cells. It is approved in the US for treating generalized pustular psoriasis (GPP) flares in adults and is under development for the treatment of Netherton syndrome.
Some of the emerging drugs for Netherton syndrome treatment, such as Sixera Pharma’s SXR1096, Daiichi Sankyo’s DS-2325, Quoin Pharmaceuticals QRX003, and others are small molecules that target KLK5, 14, and 7 knockdown or ablation. Further, their in vivo animal models and in vitro skin models incorporating human epidermal keratinocytes show promising outcomes in alleviating Netherton syndrome symptoms.

Research Advances: Progress and Promising Directions
In recent years, new advances have been made in understanding Netherton syndrome. This knowledge and the development of pathogenesis-based therapeutics, such as protein replacement therapy and gene therapy, look promising for patients suffering from Netherton syndrome. Several preclinical experiments are now underway to investigate the efficacy of these novel techniques. Significant advances in pathogenesis-based therapeutics, such as enzyme replacement therapy and gene therapy, have also been developed and may have new insights into the immunological profile of patients with inherited skin diseases. Furthermore, some case studies reveal that biological treatments may be excellent therapy options, and it is expected that some of these new medicines will demonstrate efficacy and be implemented in the treatment of Netherton syndrome.
Gene therapy to restore the function of the wild-type gene has the potential to produce positive results. Genetically corrected autologous skin grafts can be transplanted in Netherton syndrome patients. Using a viral vector to introduce the wild-type gene copy into cells is a traditional strategy. However, safety concerns about insertional mutagenesis occur when a viral vector is used. Furthermore, virus injection causes the formation of antibodies, which may impact later virus transduction. Gene therapy has not yet been explored as widely as biological. Few studies that have been published demonstrated that gene therapy could be a safe and effective option, but further investigation is essential. A classic example is Krystal Biotech’s KB104, a redosable off-the-shelf gene therapy designed to deliver two copies of the serine protease inhibitor SPINK5 to relevant skin cells when applied topically to treat Netherton syndrome. Currently, it is in the preclinical stage of development.
Another approach would be the use of replacement therapy that involves the replacement of a deficient structural protein or enzyme. For Netherton syndrome, it could be applicable for recessive forms, where a deficiency of a LEKTI1 leads to a form of ichthyosis. Azitra’s ATR-12 is a microbiome-based product designed to reside on the skin and continuously deliver the missing protein. It is a proprietary strain of Staphylococcus epidermidis engineered to express therapeutic levels of LEKTI protein to treat patients suffering from Netherton Syndrome.
Research in the field of Netherton syndrome treatment continues to evolve, and ongoing investigations are aimed at gaining a deeper understanding of the underlying molecular mechanisms, identifying potential therapeutic targets, and developing more effective treatments. Some potential future research directions include gene insights, molecular pathways, and targeted therapies to restore skin barrier functioning and inflammation in patients suffering from Netherton syndrome.
Therefore, today Netherton syndrome patients’ health and well-being are top priorities, and providing them with new, effective, and noninvasive treatment choices should be the ultimate goal. The current therapeutic strategies to re-establish the functional barrier in Netherton syndrome patients are inefficient and unspecific. Therefore, there is a strong need to develop improved and targeted therapies for patients suffering from Netherton syndrome. Few therapies are already in preclinical and clinical trials for Netherton syndrome, where they show encouraging outcomes and provide hope. With the advent of gene therapy and other advanced therapies, a paradigm shift with more ambitious treatment goals, including disease modification and potential cures, is on the horizon to treat this rare disease.




