Hemophilia A Beyond Standard Factor Therapies: Can Gene Therapy, anti-TFPI, and siRNA Siege HEMLIBRA and ALTUVIIIO’s Throne?
Aug 22, 2025
Table of Contents
Over the past four decades, hemophilia A treatment has advanced remarkably. Beginning with the introduction of cryoprecipitate in 1964, treatment progressed to plasma-derived FVIII, then to recombinant FVIII therapies in the 1980s. The 1990s and 2000s saw the rise of Standard Half-life (SHL) products, followed by Extended Half-life (EHL) therapies in the 2010s, which enabled reduced dosing frequency and improved patient outcomes.
HEMLIBRA’s Continued Dominance in Hemophilia A
The hemophilia A market has historically been fragmented, but this changed with the launch of HEMLIBRA in 2017. Approved first in the US, followed by Europe and Japan, HEMLIBRA, a bispecific antibody, quickly gained traction for both inhibitor and non-inhibitor patients, offering convenient subcutaneous dosing compared to traditional IV therapies. Its rapid uptake significantly reduced reliance on bypassing agents like NOVOSEVEN and FEIBA, especially in the inhibitor segment.
Downloads
Click Here To Get the Article in PDF

Recent Articles
- First Gene Therapy for Severe Hemophilia A; FDA Approves CellTrans’s Type 1 Diabetes Cellular The...
- Global Hemophilia Market
- Ipsen to Buy Epizyme; BioMarin’s Gene Therapy for Hemophilia; AbbVie’s Qulipta for Ch...
- Roche’s HEMLIBRA: A Game Changer in Hemophilia A Treatment Landscape
- Opportunities and Challenges for Cell and Gene Therapies
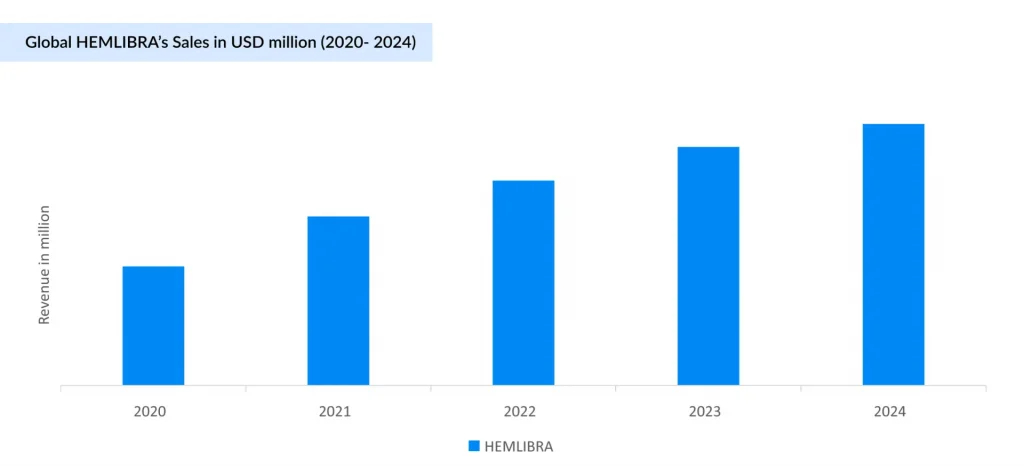
With successive label expansions (2018–2022), including for acquired and non-inhibitor Hemophilia A, HEMLIBRA has become the most prescribed therapy, overtaking ADVATE and surpassing ELOCTATE in physician preference. Backed by strong Phase III data (e.g., HAVEN 6), it has generated USD 5 billion of revenue globally (USD 3 billion in the US) in 2024, and its market share continues to rise. HEMLIBRA is now considered a first-line drug for prophylaxis in patients who have Hemophilia A with inhibitors, although ITI therapy remains the standard therapy. Additionally, HEMLIBRA may be a valuable alternative for patients who face challenges with ITI because those on HEMLIBRA prophylaxis require less frequent dosing of FVIII during ITI. The competitive landscape is intensifying, particularly for established and emerging non-factor therapies. HEMLIBRA’s growth momentum faces increasing pressure from Novo Nordisk’s Mim8, a promising bispecific antibody in development, which has about 15-fold higher potency relative to HEMLIBRA. Robust Phase III data, particularly in pediatric patients, including those with inhibitors, reinforce strong market expectations and support Novo Nordisk’s regulatory submission plans for 2025. As a result, Mim8 is well-positioned to become a key growth driver for the company within the rare disease segment in the years to come. Roche and Chugai’s NXT007 is an advanced bispecific antibody targeting Factor IXa/X, offering enhanced hemostatic activity and improved pharmacokinetics compared to current standards. Early research suggests its potential to achieve coagulation levels comparable to those of healthy individuals, which could translate into superior clinical outcomes.
HEMLIBRA has demonstrated robust market growth, taking significant share from traditional hemophilia A treatments like ADVATE and ELOCTATE. This shift is mainly driven by HEMLIBRA’S superior clinical efficacy, convenient subcutaneous administration, and high patient and physician satisfaction, reflected in its rapid uptake.
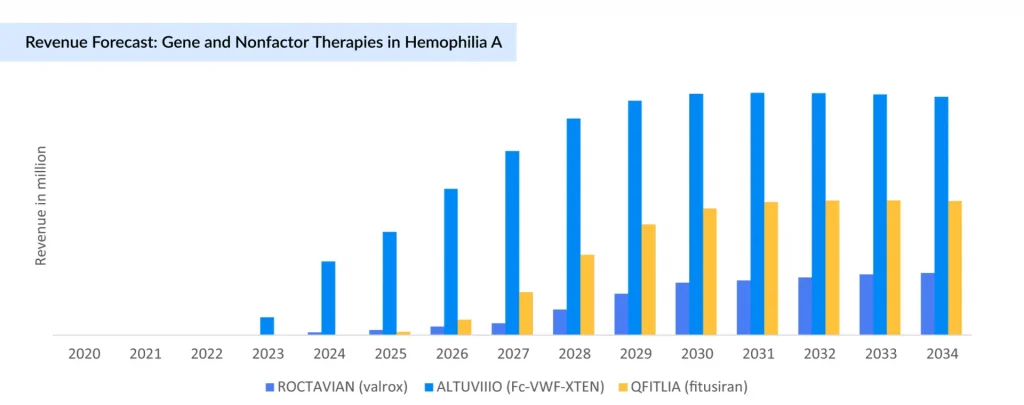
The Uphill Battle of Gene Therapy in Hemophilia A: High Potential, But Limited Traction to Date
The landscape of hemophilia A treatment has evolved significantly with the advent of gene therapies and advanced biologics developed by leading pharmaceutical companies. Foremost among these is BioMarin Pharmaceutical’s ROCTAVIAN (valoctocogene roxaparvovec), which became the first FDA-approved gene therapy for hemophilia A in 2023. Despite promising phase III results demonstrating a substantial reduction in bleeding rates and factor usage, its uptake has been cautious due to high cost, eligibility criteria, and lingering questions on long-term durability. Compared to earlier projections, ROCTAVIAN sales have been substantially lower than expected (2023: USD 3.5 million; and 2024: USD 26 million). Following this, BioMarin has adjusted its commercial strategy, narrowing its focus to the US, Germany, and Italy.
The Hemophilia A space is now characterized by a dynamic interplay of diverse therapeutic modalities competing for patient preference and payer reimbursement. Although the recent discontinuation of some clinical-stage gene therapies as Pfizer’s BEQVEZ (fidanacogene elaparvovec) and the modest real-world adoption, they underscore the profound impact that highly efficacious, convenient non-factor therapies have had on treatment paradigms. Ultimately, while gene therapies hold promise as potentially game-changing interventions, their wider adoption will depend on cost reductions and definitive long-term outcomes that can challenge the growing array of successful and more accessible treatment options. This evolving ecosystem highlights both the scientific progress and commercial complexities shaping the future of Hemophilia A management.
In December 2024, following Pfizer’s withdrawal from their global collaboration, Sangamo Therapeutics announced the reacquisition of full rights to giroctocogene fitelparvovec, an investigational gene therapy. While the termination reflects broader industry caution around gene therapy’s commercial viability, Sangamo remains committed, allocating resources to advance the program. Similarly, ASC Therapeutics’ ASC-618 and Ultragenyx’s DTX201 are positioned to deliver durable, one-time treatments with the potential to eliminate or significantly reduce the need for lifelong factor replacement or prophylaxis. Despite persistent market hesitancy, high development costs, and regulatory complexities, these companies are sustaining investment─signaling continued confidence in the long-term therapeutic and economic value of gene therapies for hemophilia A.
Sanofi/Sobi’s ALTUVIIIO on Track to Become Blockbuster Therapy in 2025
In parallel, Sanofi and Sobi have introduced ALTUVIIIO, an extended half-life recombinant FVIII replacement that combines Fc fusion and von Willebrand factor XTEN technology to prolong half-life and improve prophylaxis convenience. The initial uptake of ALTUVIIIO is strong, especially in the US market. Similar to HEMLIBRA, ALTUVIIIO is a differentiated asset that entered the market and somewhat disrupted it with a convenience narrative. ALTUVIIIO differentiates itself from the competition due to its extended half-life, which is three to four times longer than rival therapies. Growth of ALTUVIIIO is expected to be driven by patient switches from older factor replacement therapies and an increasing percentage from non-factor treatments. ALTUVIIIO recorded USD 171 million in revenue in 2023, which surged to USD 702 million in 2024, positioning it as a strong contender to become a blockbuster therapy in the hemophilia treatment landscape.
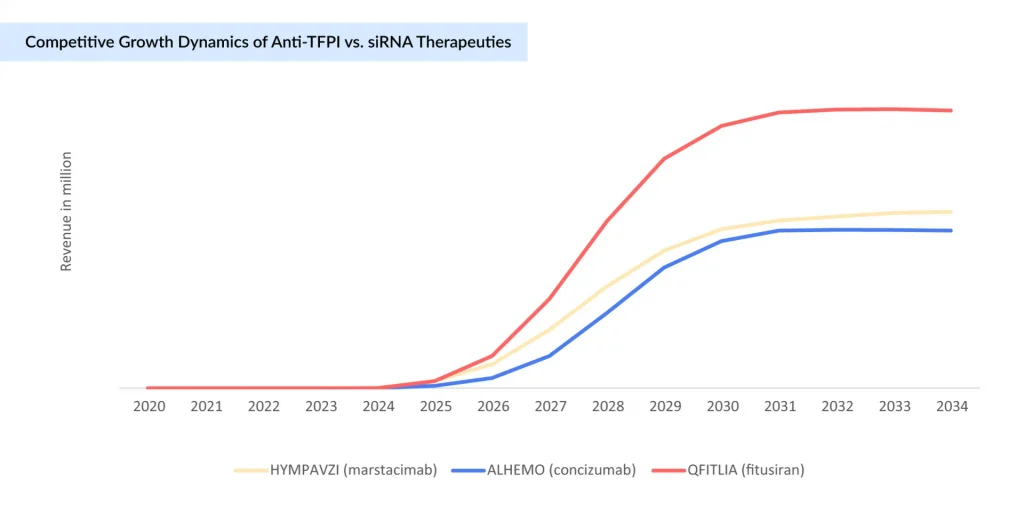
Anti-TFPI Therapies vs. SiRNA: Expanding Options Beyond Traditional Factor Replacement
Expanding beyond traditional therapies, hemophilia A care now includes two cutting-edge non-factor approaches: anti-TFPI therapies and siRNA therapies.
Pfizer’s HYMPAVZI (marstacimab), FDA-approved in October 2024 for adults and adolescents with Hemophilia A without inhibitors, has been steadily growing. It is the first once-weekly subcutaneous anti-TFPI antibody administered via a pre-filled auto-injector pen, offering a convenient and effective non-factor prophylaxis. Adoption is primarily expanding in high-income countries with established reimbursement frameworks, and ongoing pediatric studies may further extend its use. Similarly, Novo Nordisk’s ALHEMO (concizumab), an anti-TFPI subcutaneous prophylactic therapy approved earlier, is gaining traction by providing another non-factor treatment option for hemophilia A patients, including those with inhibitors. Both therapies signify a growing shift towards easier-to-administer, effective, subcutaneous treatments in hemophilia care.
siRNA therapies, led by recently approved Sanofi’s QFITLIA (fitusiran), employ RNA interference to lower antithrombin levels, rebalancing coagulation independently of FVIII and effectively controlling bleeding in hemophilia A patients, including those with inhibitors. Supported by strong clinical data showing reduced bleeding rates, it is gradually being adopted mainly in high-income countries with established reimbursement, though its high cost and monitoring requirements impact access. QFITLIA offers a convenient, effective non-factor treatment option poised to transform hemophilia care.
Both therapeutic classes represent a paradigm shift in hemophilia management by addressing significant limitations of factor replacement therapy, such as the need for intravenous dosing, frequent infusions, and the risk of inhibitor development. They expand treatment options for individuals with or without inhibitors, offering new levels of efficacy and convenience. However, both anti-TFPI and siRNA approaches still require ongoing safety monitoring, particularly to mitigate the risk of thrombotic complications.
The hemophilia A treatment market remains predominantly driven by traditional Factor VIII replacement therapies, reflecting their entrenched position and broad clinical acceptance. However, gene therapies, despite their transformative promise, have struggled to achieve significant market penetration. Several factors contribute to this slow uptake, including the high costs, patient eligibility constraints, and uncertainties around long-term durability. While gene therapies like ROCTAVIAN offer the potential for sustained benefit lasting up to eight years, their impact on market share hinges largely on addressing these economic and accessibility challenges to gain wider social acceptance.

Downloads
Article in PDF
Recent Articles
- The Evolved Gene Therapy for Hemophilia
- Ipsen to Buy Epizyme; BioMarin’s Gene Therapy for Hemophilia; AbbVie’s Qulipta for Ch...
- Hemophilia A- Market Scenario
- Top FDA Approvals to Watch in H2 2026: Regulatory Decisions Poised to Transform the Pharmaceutica...
- Cell and Gene Therapies in Rare Disorders: From Rarity to Recovery



