Roche’s HEMLIBRA: A Game Changer in Hemophilia A Treatment Landscape
Nov 07, 2022
Table of Contents
Hemophilia A treatment scenario before the launch of HEMLIBRA
Despite the ups and downs throughout history, the management of hemophilia A patients substantially improved over the past 40 years. The groundbreaking discovery of cryoprecipitate in 1964 marked the beginning of the modern progression of hemophilia treatments. A few years later, researchers found that plasma-derived factor concentrates on FVIII products. Many hemophilia A drugs that were simpler to use than cryoprecipitate or fresh-frozen plasma were approved under this category, including FEIBA (Pfizer), MONOCLATEP (CSL Behring), and HEMOFIL-M (Baxter). Recombinant therapy development in the 1980s altered the situation for people with hemophilia A. Before the approval of the first extended half-life (EHL) in the US (ELOCTATE; 2014), only short half-life (SHL) recombinant therapies were available. Other therapies are bypassing agents used specifically in hemophilia A with inhibitors. One of the highest revenue-generating bypass agents is Novonordisk’s NOVOSEVEN, which occupied the major hemophilia A market share compared with other Bypass agents such as Takeda’s FEIBA and OBIZUR (not yet approved in Japan). Out of all these therapies, the market for plasma-derived FVIII has already shrunk, and sales of SHL products have started to fall due to the acceptance and use of EHL treatments, which diminished the frequency of administration to a dismal level.
HEMLIBRA: Target patient population and strong uptake
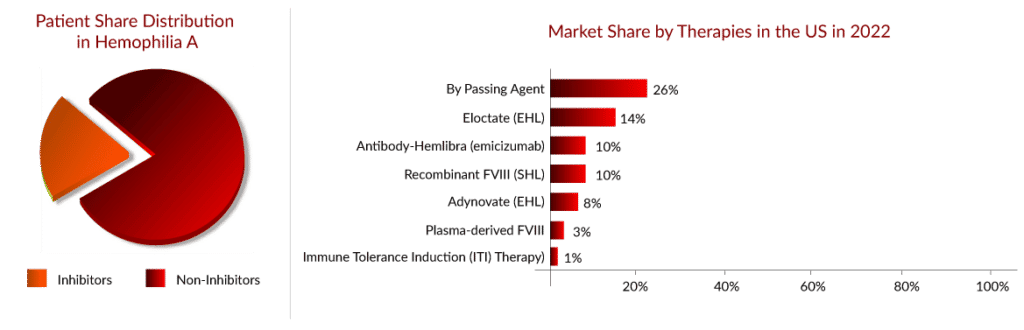
As per Delveinsight’s “Hemophilia A Epidemiology Assessment” report, there were approximately 45,000 hemophilia A cases in the seven major markets (US, EU-4, UK, and Japan) in 2021, wherein one-third of patients were the ones who developed inhibitors.
Downloads
Click Here To Get the Article in PDF

Recent Articles
- Ipsen to Buy Epizyme; BioMarin’s Gene Therapy for Hemophilia; AbbVie’s Qulipta for Ch...
- FDA Grants Priority Review to Merck’s Application for KEYTRUDA Plus Padcev; Roche and Carmot Ther...
- Roche’s HER2-Positive Breast Cancer Treatment Franchise
- Breakthrough Success: AUGMENT-101 Trial Hits Primary Endpoint with Consistent CR/CRh Rates Across...
- Allergan sell; AZ, Merck team up; Roche’s MS drug; Chinese API maker receives a warning letter
The hemophilia A market, with no single therapy controlling a significant share of the market, products have remained quite fragmented; however, the introduction of HEMLIBRA in 2017 reversed this picture. HEMLIBRA is a bispecific factor IXa- and factor X-directed antibody indicated for routine prophylaxis to prevent or reduce the frequency of bleeding episodes in adult and pediatric patients ages newborn and older with hemophilia A (congenital factor VIII deficiency) with or without factor VIII inhibitors. The first HEMLIBRA approval came in the US (November 2017), followed by Europe (February 2018) and Japan (March 2018) for hemophilia A patients with inhibitors. One of the most prescribed therapies in the inhibitor category has been bypass agents, whose market is expected to decline following the high uptake of HEMLIBRA in the inhibitor category in the coming years.
HEMLIBRA received label expansion in the US (October 2018) and Japan (November 2018) for hemophilia A without inhibitor. Roche’s HEMLIBRA continues to grow its share, overtaking Takeda’s ADVATE as physicians’ most prescribed therapy. While there are a few clear favorites (e.g., HEMLIBRA, ELOCTATE), physicians currently use a wider variety of therapies than 2 years ago. The added convenience of HEMLIBRA vs. existing FVIII replacement therapies (dosed by IV a couple of times a week) likely supports clinicians’ continued use of the therapy. While HEMLIBRA is expected to grow, precedent shows that growth has been faster than expected. HEMLIBRA is still the top-rated therapy; 57% of clinicians chose HEMLIBRA as their most preferred therapy versus the second most preferred therapy of ELOCTATE.
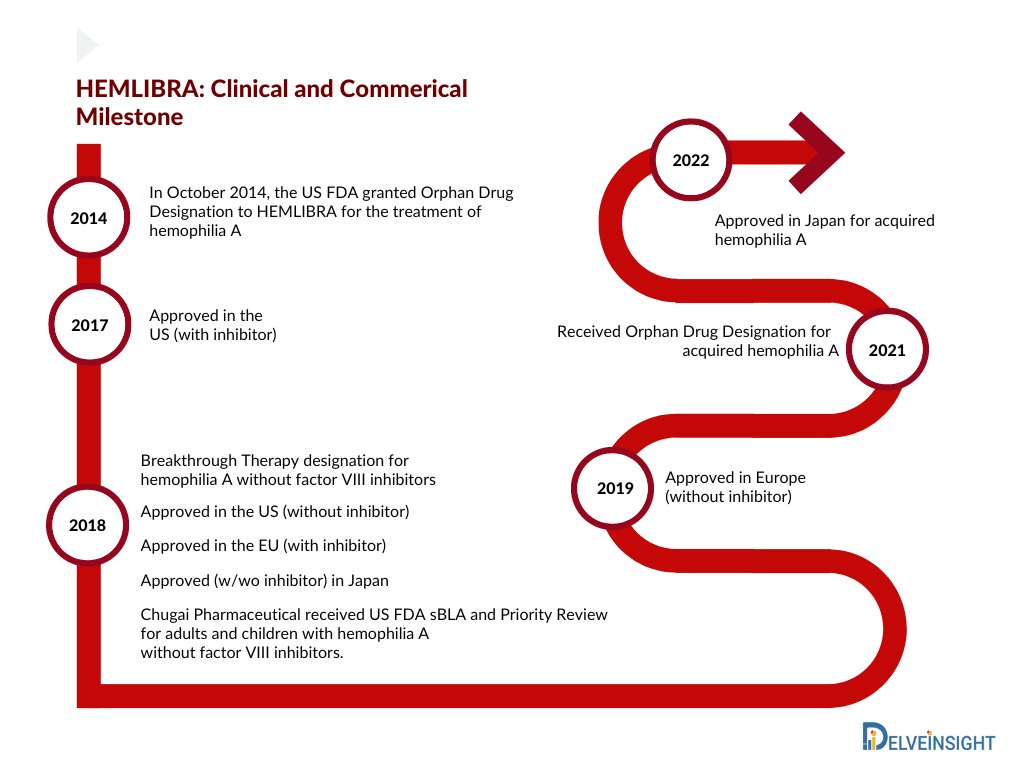
Consecutive approvals (2017, 2018, 2019, and 2022) led to an increase in the market share of HEMLIBRA. Owing to the label expansion in Europe for severe hemophilia A without factor VIII inhibitors, the uptake of HEMLIBRA increased in 2019 and captured approx. USD 1,350 million in the 7MM. Further, in June 2022, Chugai received approval in Japan for acquired hemophilia A based on a domestic Phase III (AGEHA) study. Whereas Roche is planning for a further label expansion in Europe to include mild/moderate hemophilia A without inhibitors patient’s based on Phase III data (HAVEN 6) in 2022. Therefore, we estimate Hemlibra to reach approximately USD 5,000 million by the year 2025 in the 7MM.
Patients Switching to HEMLIBRA
Since the launch of HEMLIBRA in 2018 for the non-inhibitor segment, the hemophilia A patient share has begun to switch from EHL products to HEMLIBRA, which can be easily witnessed through the strong uptake of HEMLIBRA during the last few years, where it had crossed a billion in opportunity in 2019. According to the Global Pharma survey 2022, HEMLIBRA, as of 1Q22, had 17,000 patients globally already on therapy.
Factors contributing to Patient Switching to HEMLIBRA
The patients switch to HEMLIBRA mainly based on the physicians’ recommendation; around 50–60% of physicians currently consider HEMLIBRA as the first-line hemophilia A treatment. The main reasons for patient switching and physician recommendation are the low frequency of administration (as low as once a month) and better response duration.
As per primary research, of the 21 current US hemophilia A patients surveyed, eight changed to HEMLIBRA primarily for reduced bleeds, seven for less frequent dosing, and four based on doctor recommendations. The switching source was 11 from ADYNOVATE/ADVATE, five from ELOCTATE, and two from KOVALTRY. The main reason to switch patients to HEMLIBRA was treatment burden and/or limited venous access for both inhibitor and non-inhibitor patients. Due to the high level of satisfaction with HEMLIBRA, with no current HEMLIBRA patients actively looking to switch. After switching from factor VIII prophylaxis to HEMLIBRA, adults and young adults without factor VIII inhibitors (48 people) experienced 68% fewer treated bleeds.
Upcoming competition for HEMLIBRA
While patient switching and retention rate, along with ease of administration, are the contributing factors to Hemlibra’s strong uptake, we expect a few upcoming therapies to impact the market share of Hemlibra in the near future. Novo Nordisk’s concizumab, a subcutaneously administered Tissue factor pathway inhibitor (TFPI) therapy, is expected to enter first in 2023 for patients with inhibitors, followed by its launch in 2024 in the non-inhibitor segment in the United States. Interestingly, the drug has significantly reduced bleeding rates for hemophilia A and B patients with inhibitors in a Phase III trial, based on which the company has recently filed for approval in Japan. Moving onto another therapy from the same class, Anti-TFPI, Pfizer’s Marstacimab is also expected to enter the US market initially for the non-inhibitor segment.
Following the approval of Anti-TFPI, Sanofi’s Fitusiran, a subcutaneously administered small interference RNA therapeutic, is also expected to enter the US market in 2025. Fitusiran is a siRNA designed to lower levels of antithrombin, with the goal of promoting sufficient thrombin generation to prevent bleeding. One of the key advantages of this therapy is its once/twice monthly dosing in both hemophilia A and B patients; however, we remain skeptical about the future of this drug due to its safety concerns.
In the upcoming years, it is anticipated that several additional hemophilia A therapies, including Valrox (Biomarin Pharmaceutical), BIVV001 (Sanofi), SPK-8011 (Roche/Spark Therapeutics), and giroctocogene fitelparvovec (Pfizer/Sangamo Therapeutics), will enter the 7MM and address the unmet needs of hemophilia A patients.

FAQs
Hemophilia A is a genetic bleeding disorder in which a person lacks or has low levels of clotting factor VIII proteins. Blood cannot clot properly to control bleeding if factor VIII levels are low. Even though it is passed down from parents to children, approximately one-third of cases discovered have no prior family history.
The severity and hemophilia A symptoms can vary greatly between individuals. Hemophilia A can be mild, moderate, or severe. In these categories, factor VIII levels are 5-40% of normal, 1-5%, and less than 1%, respectively.
Genetic inheritance is the cause of hemophilia A. It is caused by an X-linked recessive trait, with the defective gene on the X chromosome. Females have two X chromosome copies. So, if the factor VIII gene on one chromosome fails, the gene on the other chromosome can produce enough factor VIII.
Screening tests and clotting factor tests are used in the hemophilia A diagnosis. Screening tests are blood tests that determine whether or not the blood is clotting properly. Clotting factor tests, also known as factor assays, are required to determine the cause of a bleeding disorder. This blood test determines the type and severity of hemophilia.
HEMLIBRA is a prescription medication used for routine prophylaxis to prevent or reduce the frequency of bleeding episodes in adults and children with hemophilia A who have factor VIII inhibitors or not.
Downloads
Article in PDF
Recent Articles
- First Gene Therapy for Severe Hemophilia A; FDA Approves CellTrans’s Type 1 Diabetes Cellular The...
- FDA permits COVID-19 treatment, Roche’s antibiotics pact, Moderna’s vaccine, FDA perm...
- Reblozyl under FDA review; Audentes buyout & Tecentriq’s new approval
- FDA Approves Xolair for Food Allergies; FDA Accelerated Approval for Iovance’s AMTAGVI; Astellas ...
- Cell and Gene Therapies in Rare Disorders: From Rarity to Recovery



