What Does the Future Hold for Gene Therapy in the Duchenne Muscular Dystrophy (DMD) Treatment Market?
Aug 23, 2024
Table of Contents
Duchenne Muscular Dystrophy (DMD) is a rare disorder, but it is one of the most common genetic conditions, affecting roughly 1 in every 3,500 male births worldwide. Gene therapy for Duchenne Muscular Dystrophy (DMD) is poised to revolutionize the treatment landscape by addressing the underlying genetic cause of the disease rather than merely managing its symptoms. DMD is caused by mutations in the DMD gene, which encodes the dystrophin protein crucial for maintaining the integrity of muscle cells. The absence or dysfunction of dystrophin leads to progressive muscle degeneration and weakness, ultimately resulting in severe complications like cardiomyopathy and respiratory failure.
Gene therapy aims to introduce a functional version of the dystrophin gene or a microdystrophin gene—an engineered, smaller version that retains the essential functions of the original protein—into patients’ muscle cells. This therapeutic approach has shown promise in preclinical and early clinical trials, where it has been able to restore partial dystrophin production, stabilize or even improve muscle function, and potentially slow down the progression of the disease.
Downloads
Click Here To Get the Article in PDF
Recent Articles
- DelveInsight’s Otolaryngology based Gene Therapy Reports
- Sarepta Secures FDA Approval of Revised Prescribing Information for ELEVIDYS; Merck to Acquire Ci...
- FDA Approves Jardiance for Type 2 Diabetes; FDA Approves Pfizer’s LITFULO for Alopecia Areata; Sa...
- Reversing Ischemia with Gene therapy
- Juvenescence nets USD 100M; Sarepta DMD drug faces rejection
The potential of gene therapy to offer a one-time treatment that could dramatically alter the course of DMD is profound. Unlike traditional therapies that require lifelong administration and only target symptoms, gene therapy targets the root cause of DMD. As research advances and more clinical trials demonstrate long-term safety and efficacy, gene therapy could become the new standard of care, offering hope for significantly extended life expectancy and improved quality of life for individuals with DMD.
Duchenne Muscular Dystrophy Epidemiology
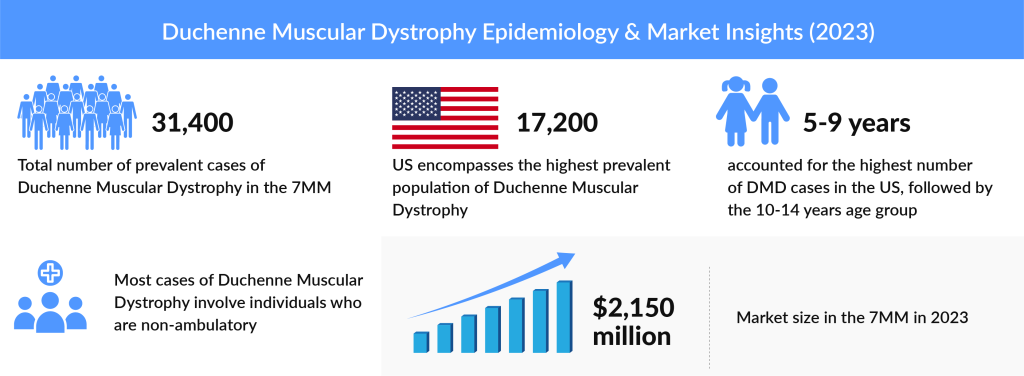
As per DelveInsight Duchenne Muscular Dystrophy Epidemiology Report, the total DMD prevalent population was more than 31K in the 7MM in 2023, which is further expected to increase by 2034. The United States accounted for approximately 17K prevalent cases of DMD in 2023 which was the maximum in the 7MM. Among the EU5 countries, the UK had the highest prevalent population of DMD, while Spain had the lowest DMD cases in 2023.
Duchenne Muscular Dystrophy Treatment Outlook
The only Duchenne Muscular Dystrophy treatments available are steroids like dexamethasone and gene-targeting therapies, including exon skipping from Sarepta Therapeutics and NS Pharma for two small subsets of patients. These DMD therapies may, at best, slow the progression of Duchenne. The Food and Drug Administration approved the therapies after studying a few dozen boys. The approved DMD therapies received a positive response and helped the patients.
Explore our blog to learn more about Duchenne Muscular Dystrophy Treatment Market
According to DelveInsight’s Duchenne Muscular Dystrophy Market research report, the total market size in the 7MM was 2.5 billion in 2023. The US accounts for the maximum portion of the global Duchenne Muscular Dystrophy treatment market. The factors driving this growth are newborn screening for DMD, increasing awareness programs, upcoming launches and approvals, and robust pipeline activity in gene therapy for DMD.
Upcoming Potential Duchenne Muscular Dystrophy Gene Therapy
Duchenne Muscular Dystrophy has long been a promising candidate for gene therapy, but overcoming several difficult technical challenges has proven difficult. For example, the dystrophin gene is too large to fit into the adeno-associated viruses, or AAVs, that are commonly used to deliver gene therapies. Researchers are trying to ensure that enough gene therapy product was delivered to muscle tissue to have an effect.
Moreover, the companies are hoping that their Duchenne Muscular Dystrophy treatment will slow or even stop disease progression, giving patients a chance to avoid the devastating effects of Duchenne.

TAS205: Taiho Pharma
TAS-205 (pizuglanstat) is a selective hematopoietic prostaglandin D synthase (HPGDS) inhibitor discovered by Taiho Pharmaceutical. It is under development as a DMD treatment which can be used regardless of the dystrophin gene mutation type, controlling the decline in motor function in DMD patients by inhibiting HPGDS, which exacerbates the inflammatory response in DMD patients’ muscles. Currently, It is in Phase III.
In 2020, an early Phase II clinical trial suggested that oral treatment with TAS-205 is safe and may help delay muscle deterioration in patients with Duchenne muscular dystrophy (DMD). The study, titled “Early Phase II trial of TAS‐205 in patients with Duchenne muscular dystrophy,” was published in the journal *Annals of Clinical and Translational Neurology*.
Prostaglandin D2 (PGD2), a hormone-like molecule produced by the enzyme hematopoietic prostaglandin D synthase (HPGDS), plays a role in inflammation and muscle tissue necrosis in DMD patients. Blocking PGD2 production could potentially reduce inflammation in these patients. TAS‐205, developed by Taiho Pharmaceutical, is an HPGDS inhibitor. In a mouse model of DMD, TAS-205 improved motor function and slowed muscle necrosis. A previous Phase I study (NCT02246478) in DMD patients showed that TAS-205 reduced PGD2 levels in urine.
The Phase II trial (NCT02752048), sponsored by Taiho, evaluated the efficacy and safety of TAS‐205 in male DMD patients aged 5 and older in Japan. The study included 35 patients who received either TAS-205 or a placebo orally. Eleven patients received a low dose (6.67 to 13.33 mg/kg), 12 received a higher dose (13.33 to 26.67 mg/kg), and the remaining 12 received a placebo. The average age of participants across all groups was approximately 8 years.
PF-06939926: Pfizer
Pfizer’s PF-06939926 is an investigational gene therapy for Duchenne Muscular Dystrophy treatment. It is a recombinant adeno-associated virus serotype 9 (AAV9) capsid containing a shortened version of the human dystrophin gene (mini-dystrophin) controlled by a human muscle specific promotor. Because of its ability to target muscle tissue, the AAV9 capsid was chosen as the delivery mechanism and is administered intravenously.
Pfizer’s PF-06939926 was designated as an Orphan Drug and Pediatric Rare Disease by the FDA in May 2017 and an Orphan Medicinal Product Designation by the EMA for the treatment of DMD. The company announced in October 2020 that its gene therapy product had also received Fast Track designation from the FDA.
But unfortunately, a participant in Pfizer’s Phase Ib open-label study died unexpectedly. The FDA has ordered a clinical halt to the trial, and Pfizer is investigating the causes of death. The patient was a part of the study’s non-ambulatory arm. Pfizer is also conducting a Phase III study of the same product, which is being developed globally. PF-06939926 is among the two gene therapies in late-stage development for DMD, with Sarepta Therapeutics’ SRP-9001 serving as its main competitor. Following this major safety event, the uncertainty surrounding PF-06939926’s future could potentially pave the way for Sarepta’s continued dominance in the field.
In June 2024, Pfizer announced that its Phase III CIFFREO study of fordadistrogene movaparvovec (PF-06939926) failed to meet its primary endpoint. The company reported that the gene therapy’s safety profile was deemed “manageable,” with adverse events primarily mild to moderate, and serious adverse events responding well to clinical management. This follows a recent fatality in the Phase II DAYLIGHT study, which led to a dosing pause in CIFFREO. Meanwhile, Pfizer’s other gene therapy, fidanacogene elaparvovec-dzkt (Beqvez), was approved by the FDA for hemophilia B treatment on April 26, 2024, and by Health Canada in January 2024. Beqvez is indicated for adults with moderate to severe hemophilia B who have specific clinical needs and do not possess neutralizing antibodies to the AAVRh74var capsid.
SGT-001: Solid Biosciences
SGT-001 is a novel AAV vector-mediated gene transfer therapy that aims to address the underlying genetic cause of DMD. SGT-001 is a systemically administered candidate that provides the body with a synthetic dystrophin gene called microdystrophin. This microdystrophin encodes a functional protein surrogate expressed in muscles and helps stabilize essential associated proteins such as neuronal nitric oxide synthase (nNOS). According to data from Solid’s clinical program, SGT-001 has the potential to slow or stop the Duchenne progression, regardless of genetic mutation or disease stage,
SGT-001 is based on groundbreaking dystrophin biology research conducted by researchers at the University of Washington and the University of Missouri. SGT-001 has received Rare Pediatric Disease and Fast Track Designation in the United States and Orphan Drug Designation in the US and EU in 2017.
In September 2021, the company reported Positive 1.5-year functional data and patient-reported outcome measures (Pediatric Outcomes Data Collection Instrument, or PODCI) for Patients 4-6 in the ongoing IGNITE DMD Phase I/II clinical trial of SGT-001. The company previously reported 1-year data for the same measures in March 2021. The findings showed that the microdystrophin protein remains expressed and functional in biopsy samples collected 12 to 24 months after SGT-001 administration. These findings showed a significant improvement in patient-reported outcomes and provided encouraging evidence of functional benefit 1.5 years after treatment when compared to natural history data.
In April 2024, the FDA granted SGT-003 rare pediatric disease status. This designation is awarded to therapies with the potential to address rare diseases primarily affecting children and adolescents with DMD. It could enable Solid to qualify for a priority review voucher upon seeking approval for another experimental treatment, which the company may also sell or transfer. This recognition comes on the heels of the FDA’s recent orphan drug designation for SGT-003, further supporting its potential as a significant advancement in DMD treatment.
RGX-202: Regenxbio
RGX-202 is intended to deliver a transgene encoding a novel microdystrophin with functional elements of the C-Terminal (CT) domain found in naturally occurring dystrophin. In preclinical studies, the presence of the CT domain was shown to recruit several key proteins to the muscle cell membrane, resulting in improved muscle resistance to contraction-induced muscle damage in dystrophic mice.
Additional design elements, such as codon optimization and CpG content reduction, have the potential to enhance gene expression, increase translational efficiency, and reduce immunogenicity. The NAV AAV8 vector, which has been used in numerous clinical trials, and a well-characterized muscle-specific promoter (Spc5-12) are used in RGX-202 to support the delivery and targeted expression of genes throughout skeletal and heart muscle.
In January, The FDA approved Regenxbio’s request to conduct a Phase I/II clinical trial in the United States to assess the safety and efficacy of RGX-202, its experimental gene therapy for Duchenne Muscular Dystrophy (DMD).
The Phase, I/II trial, named AFFINITY DUCHENNE study, which is set to begin in the coming months. Contact information and locations are not yet available, but initial trial sites are expected to open in the United States, with sites in Canada and Europe to follow.
In November 2021, the FDA designated RGX-202 as an orphan drug for the treatment of Duchenne Muscular Dystrophy. This designation is designed to provide regulatory assistance, financial benefits to the therapy’s clinical research and evaluation, and a seven-year marketing exclusivity period in the United States after regulatory clearance.
In April 2023, REGENXBIO secured fast-track status for RGX-202, its AAV therapy aimed at delivering a transgene for a novel microdystrophin to address the muscle degeneration associated with Duchenne muscular dystrophy (DMD). This regulatory privilege, alongside the previously granted orphan-drug and rare pediatric disease designations, enables REGENXBIO to benefit from more frequent interactions with the FDA and potentially a priority review if RGX-202 progresses to an approval filing. The FDA’s clearance for a Phase I/II trial, set to deliver initial data in the second half of the year, marks a significant milestone in advancing this one-time DMD treatment.
In August 2024, REGENXBIO Inc. announced new, positive interim safety and efficacy data from the Phase I/II AFFINITY DUCHENNE® trial, evaluating RGX-202 in patients aged 1 to 11 years with Duchenne muscular dystrophy. These findings represent a significant step forward in the potential treatment of Duchenne, underscoring the promise of RGX-202 in addressing this challenging condition.
What Lies Ahead?
Gene therapy for Duchenne Muscular Dystrophy is to be the most promising DMD pipeline candidate in the market. Sarepta is a market leader in this category, with three out of every five marketed therapies in the US market addressing DMD.
Sarepta and Pfizer are evaluating their lead candidates for DMD gene therapy in the late stages. However, unlike Sarepta, Pfizer does not have any additional candidates that may join the market and earn market share if its DMD gene therapy treatment fails to win approval, implying that the stakes are higher for the latter.
Even if both gene therapies reach the market, PF-06939926 is likely to face a delay due to the recent death in its Phase Ib trial. As a result, SRP-9001 would gain a competitive edge. Regardless, Pfizer will need to examine the situation and acquire the data necessary to continue the Phase Ib trial and make changes to future trials, such as omitting certain mutation types.
Despite the risks mentioned above, which may result in lower uptake than Sarepta’s product, Pfizer could still capture a significant market share and see a return on its investment before more DMD gene therapies enter the market.

Downloads
Article in PDF
Recent Articles
- Watershed Moment for Cell Therapies and Complicated Journey of Gene Therapies in Japan
- Avrobio’s Gene Therapy for Cystinosis Treatment: A New Hope to Overcome the Existing Medical Unme...
- DelveInsight’s Otolaryngology based Gene Therapy Reports
- DelveInsight’s Immunology based Gene Therapy Reports
- DelveInsight’s Ophthalmologic disorders based Gene Therapy Reports



