Labyrinth of LDL-C: Navigating Challenges in Homozygous Familial Hypercholesterolemia Treatment
Aug 25, 2023
Table of Contents
Homozygous familial hypercholesterolemia (HoFH) is a rare form of familial hypercholesterolemia, an autosomal-dominant genetic disorder of lipid metabolism characterized by strikingly elevated levels of low-density lipoprotein cholesterol (LDL-C).
Heterozygous Familial Hypercholesterolemia vs. Homozygous Familial Hypercholesterolemia
Heterozygous familial hypercholesterolemia individuals inherit one mutated copy of the LDL receptor gene from one parent and one normal copy from the other parent. This means they have one faulty gene and one functional gene resulting in a less severe form. While most people suffer from HeFH, in rare cases, the mutations in both copies of genes related to cholesterol metabolism give rise to the lethal form of familial hypercholesterolemia, i.e., HoFH. A major difference between homozygous and heterozygous familial hypercholesterolemia is the presence of cholesterol levels. HeFH patients can have cholesterol levels in the 350–550 mg/dL range, while HoFH can be in the 650–1,000 mg/dL range. DelveInsight epidemiology analysis shows more than 1,300 individuals in the US were diagnosed with HoFH in 2022.
Downloads
Click Here To Get the Article in PDF

Recent Articles
- Takeda and AC Immune’s Alzheimer’s Deal; Eli Lilly’s Donanemab FDA Review; Bristol Myers Sq...
- A CRISPR WAY TO USE STEM CELLS
- Snippet : Genetic Mutation
- CRISPR to cure sickle cell; FDA rejected; Stem cells use; Celgene spinoff
- Most Promising Artificial Intelligence Applications in the Healthcare Segment
What is Homozygous Familial Hypercholesterolemia (HoFH)?
HoFH, a life-threatening disease, is clinically characterized by plasma cholesterol levels >13 mmol/L (>500 mg/dL), corneal arcus, and extensive xanthomas with a risk of premature and recurrent coronary events and atherosclerotic cardiovascular disease (ACVD). These clinical features of HoFH generally appear early in life, either in the first or second decade. HoFH compromises the liver’s ability to remove LDL, commonly known as “bad” cholesterol leading to high levels of LDL. These high LDL levels, if untreated, can cause aggressive narrowing and blocking of the blood vessels even before birth, which progresses rapidly, triggering serious cardiac issues for patients much earlier in life than the general population.
Mutations in four major genes have been identified to cause HoFH, including Low-density lipoprotein receptor (LDLR), Apolipoprotein B (APOB), Proprotein convertase subtilisin/kexin type 9 (PCSK9), and Low-Density Lipoprotein Receptor Adaptor Protein 1 (LDLRAP1). More than 90% of HoFH cases result from a mutation in the LDLR gene. According to the National Institutes of Health (NIH), patients with HoFH have LDL-C levels three to six times higher than normal. While in European countries, cholesterol levels are measured in mmol/L, in the US, cholesterol levels are measured in mg/dL.
Diagnostic Approaches for Homozygous Familial Hypercholesterolemia
The diagnostic journey of HoFH involves the examination of family history for clues, assessing physical manifestations like xanthomas, and delving into sophisticated genetic analyses to detect the significant mutations in LDL receptor genes. Further, once a diagnosis is established, systematic cascade screening is essential to identify family members with familial hypercholesterolemia, given the autosomal-dominant inheritance pattern of the disease.The diagnosis of HoFH can be made based on clinical and/or genetic criteria endorsed by the European Atherosclerosis Society (EAS) that require a genetic confirmation of two mutant alleles of the LDLR, APOB, PCSK9, or LDLR adaptor protein 1 gene locus; or an untreated LDL-C >500 mg/dL or treated LDL-C ≥300 mg/dL together with either cutaneous or tendon xanthoma before 10 years of age or untreated elevated LDL-C levels consistent with HeFH in both parents. However, many diagnosed cases sometimes have negative genetic testing leading to confusion and delayed treatment. Nevertheless, the American Heart Association (AHA) has addressed this challenge by encouraging the screening for elevated levels of lipoprotein(a) (Lp[a]), which is highly prevalent among patients with HoFH.
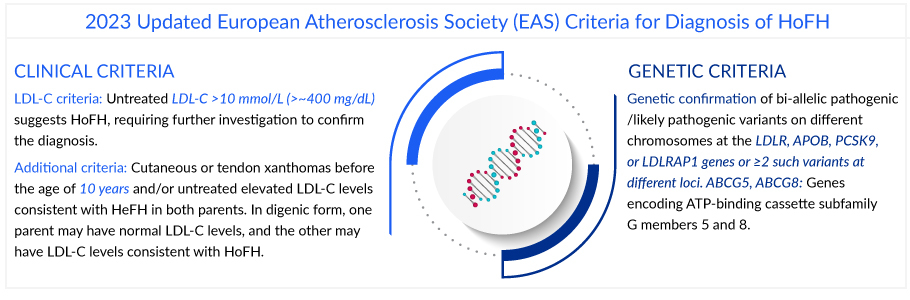
The European Atherosclerosis Society (EAS) recommends promptly referring patients with suspected HoFH to specialist centers for a comprehensive ACVD evaluation and clinical management.
Current Homozygous Familial Hypercholesterolemia Treatment Landscape
After a successful diagnosis of HoFH, patients are referred to a lipid specialist for advanced management of the condition. The homozygous familial hypercholesterolemia treatment is challenging due to the severe elevation of LDL-C levels and the increased risk of early-onset cardiovascular complications and requires a combination of lipid-lowering medications, lifestyle modifications, and in some cases, advanced interventions. Reducing mortality and preventing ASCVD progression is the primary goal of homozygous familial hypercholesterolemia treatment.
According to most of the guidelines, management of HoFH should start with conventional statin therapy, and depending on the response, combination therapy should be added, and/or non-pharmacologic interventions should be considered. Statins (simvastatin, rosuvastatin, and atorvastatin) and ezetimibe are the first-line therapy for patients with HoFH. Statins reduce LDL-C by diminishing hepatic cholesterol synthesis, acting on the 3-hydroxy-3-methylglutaryl coenzyme-A (HMG-CoA)-reductase. This leads to the upregulation of hepatic LDLR expression and the increased uptake of circulating LDL particles with consequent higher biliary cholesterol excretion in the feces. Despite the first line of therapy, PCSK9 inhibitors (alirocumab and evolocumab), anti-Apo-B therapies (lomitapide), and ANGPTL3 inhibitors (evinacumab), and others are recommended. Evolocumab and alirocumab in homozygous familial hypercholesterolemia are indicated in patients with elevated LDL-C despite statins and ezetimibe therapies. Further, evinacumab for homozygous familial hypercholesterolemia, unlike PCSK9 inhibitors, provides robust LDL-C reduction (−43.4%) even in patients with LDLR null variants. However, its impact on cardiovascular events and long-term effects has not been determined and needs evaluation.The American Heart Association/American College of Cardiology recommends initiating a high-intensity statin to lower LDL-C levels by a minimum of 50% in high-risk and very high-risk patients. The European Atherosclerosis Society guidelines state that goal LDL-C levels in patients with HoFH are <100 mg/dL for adults without clinical ASCVD, <70 mg/dL for adults with clinical ASCVD, and <130 mg/dL in children.

Besides these interventions to lower LDL independent of LDL-receptor lipoprotein, apheresis, and liver transplantation are recommended. Lipoprotein apheresis is the most effective means of lowering LDL-C levels in patients with HoFH. It also reduces inflammatory markers, oxidative stress, and thrombogenic factors and improves endothelial functions. On the contrary, liver transplantation is considered a curative treatment or last resort for HoFH, with substantial LDL-C reductions of up to 80%.
Barriers to the Management of HoFH Patients
Substantial improvements in treating familial hypercholesterolemia have been achieved over the last 25 years with statins, ezetimibe, VLDL synthesis inhibitors, and PCSK9 antibodies. Unfortunately, many HoFH patients still do not achieve targeted LDL-C levels. The major challenges in managing HoFH include:
- Diagnostic gaps: HoFH is often not diagnosed until late childhood or adolescence, which delays the implementation of appropriate treatment. Further, there is a lack of national cholesterol screening programs and limited accessibility to genetic testing. Other obstacles to early diagnosis include the limited public awareness of familial hypercholesterolemia as a disease entity and its inherited nature.
- Severe hypercholesterolemia: HoFH patients experience extremely high LDL cholesterol levels from birth, which may be four to five times higher than in the general population, making it difficult to reach optimal LDL-C levels. Lowering these levels to acceptable ranges is a persistent challenge, often requiring aggressive and unconventional homozygous familial hypercholesterolemia treatment approaches.
- Sub-optimal therapies: Statin intolerance leads to discontinuation of treatment or dose reduction, which places patients with increased LDL-C levels at risk of cardiovascular events. Further, the patients with HoFH lack fully functioning LDLR, and statins are not generally effective in this population, in whom only modest reductions in LDL-C levels of ~20 % are observed. Other approved therapies are limited due to their side effects; JUXTAPID is restricted by its potential to induce elevation of liver enzymes, hepatic steatosis, and patient toleration due to GI side effects and failure to achieve targeted LDL-C level. Further, many therapies have not been adequately studied in the pediatric population.
- Treatment adherence: The complex homozygous familial hypercholesterolemia treatment regimens, including frequent apheresis sessions, injections, and medications, can be overwhelming and impact patient adherence. Balancing these demands with daily life can be intimidating.
- Long-term cardiovascular risk management: HoFH patients remain at high risk for cardiovascular events despite aggressive management. Untreated individuals with familial hypercholesterolemia have a 20-fold increased risk for coronary artery disease. Untreated men have a 50% risk of a nonfatal or fatal heart attack by age 50; untreated women have a 30% risk by age 60. Further, limited data on cardiovascular disease risk in patients with familial hypercholesterolemia is available as a hurdle in treatment.
- Limitation of lipid apheresis: Although lipid apheresis is considered the most effective way of lowering LDL-C, being an intermittent therapy, it leads to a saw-tooth-like pattern of LDL-C levels. Therefore, the extent of the rebound of the LDL-C levels may diminish the expected benefits of apheresis therapy. Further, the semi-invasive, time-consuming, and chronic nature of lipid apheresis significantly contributes to the high refusal and low adherence rates.
- Drawbacks of liver transplant: Despite being a curative option, its use has been limited due to several drawbacks, including the scarcity of donors, the high risk of surgical complications, and the life-long requirement of immunosuppressive therapy to prevent graft rejection. Further, the LDL-C level is not guaranteed even after a liver transplant, and patients must take lipid-lowering therapies.
- Challenges in clinical trials: Due to the rarity of HoFH, research is limited compared to other prevalent disorders. Limited drugs for HoFH have been investigated in clinical trials, and only a few have been successful. Difficulty recruiting patients, high cost, and long follow-up periods are the major challenges a homozygous familial hypercholesterolemia clinical trial faces.
HoFH patients are always at high risk of cardiovascular-related complications and premature death, and yet they remain underdiagnosed and undertreated. More research and homozygous familial hypercholesterolemia clinical trials are needed to explore new therapeutic approaches that can effectively lower the LDL-C to the targeted levels and fulfill these anesthetic challenges.
Emerging HoFH Treatment Options
While there are HoFH treatments available, they may not be universally accessible or affordable for all patients and are associated with side effects. Nevertheless, the advances in research have led to the discovery of novel molecules like small interfering RNA (ARO-ANG3 and LEQVIO [inclisiran]) and recombinant fusion protein (lerodalcibep [LIB003]) that may offer potential options to lower LDL significantly.
Novartis and Alnylam Pharmaceuticals’ LEQVIO (inclisiran) is a novel PCSK9 inhibitor being developed for treating HoFH. It blocks the translation of PCSK9 messenger RNA, leading to its degradation by the RNA-induced silencing complex, thereby decreasing intrahepatic and plasma PCSK9 concentrations. Compared with other PCSK9-mAbs, inclisiran has a more convenient dose regimen of 300 mg twice-yearly injections and could be useful for enhancing patient compliance. It is already approved for HeFH, and there are signs that LEQVIO may surpass PCSK9 rivals, including REPATHA and PRALUENT.
Lerodalcibep (LIB003), developed by LIB Therapeutics, is a Chinese hamster ovary-cell line-derived recombinant fusion protein therapeutic agent with a PCSK9-binding domain and human serum albumin (HSA). The small size (77 kD) and high solubility of LIB003 allow for a smaller SC injection volume than achievable with monoclonal antibodies and impart a stable and prolonged duration of LDL-C reduction between injections.
Arrowhead Pharmaceuticals’ ARO-ANG3 potentially inhibits ANGPTL3 protein lowering the serum LDL level. The drug addresses the need for additional therapy with a mechanism working other than the LDL receptor and offers a convenient dosage that improves adherence and compliance.
The upcoming drugs for HoFH treatment can potentially overcome drawbacks like compliance and adherence to treatment by providing a convenient dosage regimen. However, their long-term efficacy in lowering LDL-C levels remains to be validated.
Besides the above-mentioned HoFH therapies, techniques such as gene transfer, silencing, and editing are currently under investigation for HoFH. Verve Therapeutics is developing an early-stage product, VERVE-201, using CRISPR-based editing therapy.
According to DelveInsight analysts, the total homozygous familial hypercholesterolemia market size of the seven major markets (the United States, Germany, France, Italy, Spain, the United Kingdom, and Japan) is likely to cross the USD 100 million mark by 2032 owing to the launch of emerging therapies, increased awareness, and improved diagnosis.
Although there have been tremendous advancements in HoFH, several unmet needs are yet to be fulfilled. Increasing the awareness of familial hyperlipidemia among healthcare practitioners will ultimately lead to better prevention of HoFH. Due to cardiovascular and atherosclerotic risks, diagnosing as early as possible and appropriate treatment initiation is necessary. Further, most emerging and marketed therapies are add-on therapy, so the necessity to develop effective first-line therapy prevails.
FAQs
HoFH is a rare and severe genetic disorder that affects cholesterol metabolism. It is an inherited condition caused by mutations in both copies of the (low-density lipoprotein receptor) LDLR gene or other genes related to cholesterol metabolism, such as the APOB or PCSK9 genes.
The hallmark symptom of HoFH is severely elevated LDL-C levels from birth, often ranging well above 500 mg/dL. This excessive cholesterol buildup in the blood leads to the premature development of atherosclerosis and cardiovascular disease. Individuals with HoFH are at high risk of suffering from heart attacks, strokes, and other cardiovascular complications at a young age, sometimes even during childhood or adolescence. Xanthomas, which are fatty deposits beneath the skin, tendons, and eyelids, may also develop in individuals with HoFH.
The diagnosis involves a combination of clinical evaluation, lipid profile testing, and genetic testing. Due to its extreme rarity and potentially severe consequences, early and accurate diagnosis is crucial for timely intervention and management. Treatment typically involves a combination of lipid-lowering medications, lifestyle modifications, and in some cases, advanced interventions.
Current treatment strategies for HoFH include a combination of high-intensity statins, cholesterol absorption inhibitors, and lipid-lowering injections such as mipomersen and lomitapide. In some cases, patients might also require apheresis, a procedure that filters LDL cholesterol from the blood.

Downloads
Article in PDF
Recent Articles
- Inozyme Raises $67M; Shanghai Miracogen partners with Synaffix; EdiGENE raises $15M; Antengene an...
- Notizia
- Most Promising Artificial Intelligence Applications in the Healthcare Segment
- Social Behavior Loss Observed in Gene-edited ants through CRISPR
- Unraveling the Potential of CRISPR Technology in the Gene-editing Space



