Revolutionizing Dilated Cardiomyopathy Treatment: The Road Ahead
Jun 24, 2024
Table of Contents
Quick facts
- The primary cause of DCM is understood to be genetic, leading to a focus on tailoring treatments based on the specific molecular origin. Genetic testing plays a crucial role in DCM, as numerous studies and case reports have demonstrated, serving as a vital tool for diagnosis, prognosis, familial screening, and reproductive decision-making.
- As an alternative to oral small-molecule therapies for DCM associated with mutations in different sarcomere-related genes, new therapies are emerging that involve stem cell transplantation and gene editing techniques. These innovative treatments focus on targeting specific gene variants that affect sarcomeric and cytoskeletal proteins and aim to address the underlying causes of DCM and enhance cardiac function.
- As emerging therapies become available, it is predicted that approximately two-thirds of patients newly diagnosed with DCM will see significant heart function improvement within the first year, with many potentially achieving full symptom resolution and normal heart function.
According to DelveInsight’s estimates, in 2023, there were approximately 554K total diagnosed prevalent cases of DCM in the US. These cases are expected to increase by 2034, attributed to several interrelated factors, ranging from genetic predispositions to lifestyle and environmental influences.
Most cases of dilated cardiomyopathy have a slow and insidious onset and may be asymptomatic in the early stages. DCM exhibits diverse clinical presentations among patients, primarily driven by a broad spectrum of genetic and non-genetic factors.
Downloads
Click Here To Get the Article in PDF

Recent Articles
- The Regenerative Medicine Revolution: Opportunities Shaping the Next Decade
- Microbial Fermentation Technology: Revolutionizing Biotechnology and Industry
- Takeda and AC Immune’s Alzheimer’s Deal; Eli Lilly’s Donanemab FDA Review; Bristol Myers Sq...
- Labyrinth of LDL-C: Navigating Challenges in Homozygous Familial Hypercholesterolemia Treatment
- CRISPR proves to be a blessing for LASSA fever
“Before we started studying DCM, we called it idiopathic myopathy as we were not sure what caused it. It ran in families; the response to myosin activators varies depending on the underlying genetic etiology. Therefore, we believe DCM is a complex disease with multiple genetic causes, and identifying the underlying genetic etiology can help tailor treatment strategies to individual patients.”
– Ohio State Wexner Medical Center
The most common clinical presentations of DCM include arrhythmia and heart failure, which can manifest as palpitation, excessive sweating, ankle swelling, progressive dyspnea, and fatigue following mild exercise. In advanced stages, patients may also experience additional symptoms such as abdominal distension, loss of appetite, edema, pleural effusion, and ascites.
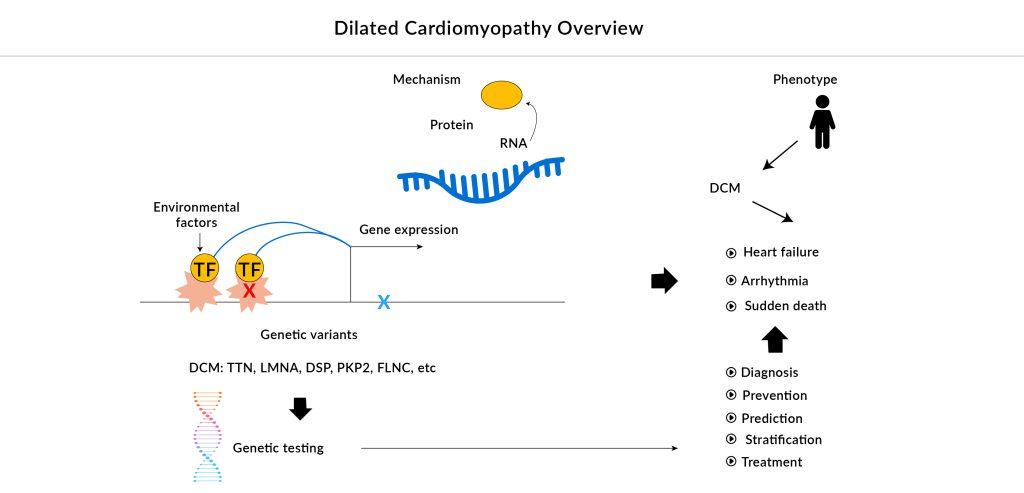
The primary goals of current DCM treatments are to enhance cardiac function and reduce the symptoms of heart failure. Angiotensin-converting enzyme (ACE) inhibitors, angiotensin II receptor blockers (ARBs), beta-blockers, aldosterone antagonists, and diuretics are important pharmacological treatments. Patients with DCM are advised to use ACE inhibitors and ARBs because they are particularly beneficial for heart failure with a lower ejection fraction. Sometimes, beta-blockers such as carvedilol, metoprolol, propranolol, and atenolol can lessen heart enlargement. However, side effects from beta-blockers can include depression, lethargy, poor academic performance, dizziness, bradycardia, low blood pressure, and fluid retention. The choice of anticoagulants, including aspirin and dipyridamole, is based on the likelihood of blood clot formation. In addition, arrhythmias are commonly treated with anti-arrhythmic medications such as lidocaine, procainamide, and amiodarone.
The only FDA-approved drug in its class, CORLANOR (ivabradine) from Amgen, is a cyclic nucleotide-gated channel blocker that is activated by hyperpolarization. It lowers heart rate by selectively inhibiting the IF current, which decreases the spontaneous pacemaker activity of the cardiac sinus node without affecting ventricular repolarization or myocardial contractility. In April 2019, the US FDA approved ivabradine for treating stable symptomatic heart failure resulting from dilated cardiomyopathy in pediatric patients aged 6 months to 18 years. Although ivabradine has been effective in reducing hospitalization rates and alleviating symptoms, it does have some limitations, such as the potential to cause bradycardia. It is mainly prescribed for patients with stable heart failure and may not be as effective for those with advanced disease who need more intensive treatment.
While current dilated cardiomyopathy treatments focus on controlling symptoms and improving heart function, there is a need for more innovative approaches to address the underlying causes of DCM and improve patient outcomes.
Addressing the gaps in dilated cardiomyopathy treatment
Despite progress in pharmacological treatments globally, there are very few novel treatments for familial DCM. Other than mutations in the TTN gene, certain genetic mutations that impact the synthesis of sarcomeric and cytoskeletal proteins are frequently responsible for the advancement of DCM. Current dilated cardiomyopathy therapies do not address these particular genetic factors. New treatments that focus on the underlying disease mechanisms of each genetic subgroup are scarce. Precision medicine approaches are urgently needed. The only significant candidates in advanced phases of development for familial DCM are danicamtiv and BC007. Late-stage results are essential to confirm their efficacy and safety, making it difficult to predict their market potential.
Although the early-stage pipeline exhibits greater activity, with several molecules in the preclinical and discovery stages, these will take time to develop. As the present pipeline is insufficient, there is a need to increase disease awareness, validate the effectiveness of current treatments, and develop additional drugs to expand the market.
A step toward personalized medicine in dilated cardiomyopathy treatment
About 40% of DCM cases are hereditary in nature. The progression of DCM to heart failure is marked by worsening wall thinning, fibrosis, arrhythmias, dyspnea, exercise intolerance, and sometimes sudden death. Accurate phenotyping of DCM patients is vital for identifying specific subgroups and tailoring treatments to individual needs. This involves comprehensive assessments of cardiac function, structure, and remodeling, as well as genetic and biomarker analyses.
Personalized medicine approaches focus on customizing dilated cardiomyopathy treatments for individual patients based on their unique genetic profiles, disease mechanisms, and therapy responses. These may include gene therapy, gene editing, and targeted treatments. This approach offers several advantages, including improved efficacy and reduced side effects. Personalized dilated cardiomyopathy treatment can be designed to target specific genetic mutations or disease mechanisms, increasing the likelihood of effective therapy. Additionally, personalized dilated cardiomyopathy treatment can help identify patients who are most likely to benefit from a particular therapy, reducing the risk of ineffective or harmful treatments as well as more precise monitoring and adjustment of treatment, enabling clinicians to optimize therapy over time.
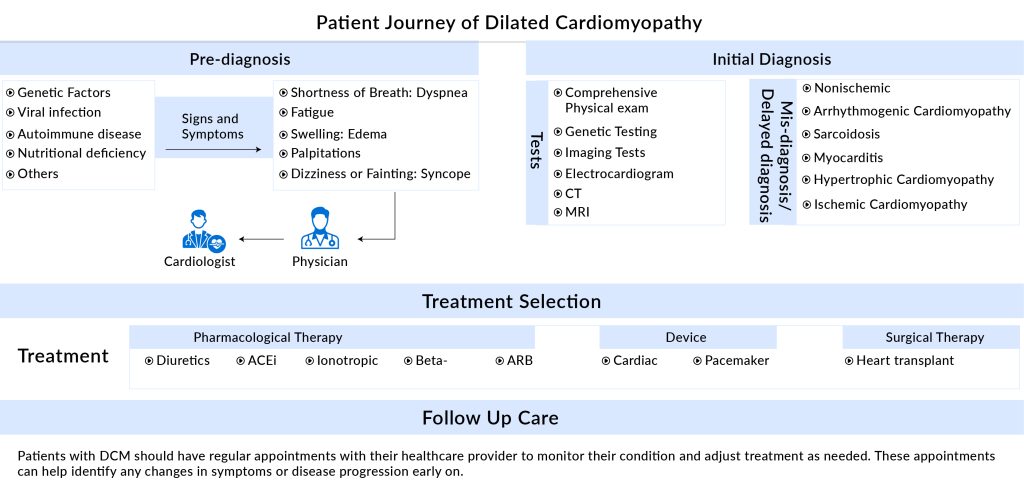
Thus, navigating the challenging path toward a personalized approach for DCM offers new hope for enhancing patient outcomes and mitigating the impact of this debilitating disease.
Upcoming therapies to improve DCM treatment
The emerging treatment strategies for DCM are based on regenerative medicine approaches, such as stem cell therapy and gene therapy that have the potential to repair and restore myocardia functioning in patients.
Gene modification
Gene therapy, involving the introduction of foreign genes into cells, has shown promise in treating diseases like DCM and inherited disorders. Adeno-associated virus (AAV) vectors have shown promising results in delivering genes to the myocardium while addressing concerns related to immunogenicity and safety. Notable preclinical programs leveraging AAV gene therapy include Rocket Pharmaceutical’s REN-001, Nuevocor’s NVC-001, and Tenaya Therapeutic’s DWORF Gene Therapy Program, among others.
Myosin Heavy Chain as a Drug Target
Myosin heavy chain, a key protein in cardiac sarcomere, is a potential drug target for DCM patients. Bristol Myers Squibb’s danicamtiv (MYK-491) recruits myosin motors and increases myofilament Ca2+ sensitivity, targeting biomechanical defects and improving cardiac contractility in DCM patients. Recently the company terminated its Phase II trial for DCM in the US, Spain, Germany, and the United Kingdom citing change in business objectives as the reason.
The use of myosin activators in DCM is under scrutiny, with concerns about their effectiveness in genetic progression. Clinical trials show some benefits in human heart failure, but the population of patients with genetic DCM is unclear. Moreover, the potential benefits of combining myosin activators with other therapies, such as beta-blockers or angiotensin-converting enzyme inhibitors, need to be explored to optimize treatment outcomes.
CRISPR-to activate expression of TTN
CRISPR activation of the TTN gene has shown promise in reversing haploinsufficiency and addressing functional deficits caused by TTN truncation variants, a common genetic cause of DCM. This method targets the genetic mutation responsible for DCM, offering a more precise and effective treatment compared to traditional nonspecific approaches. However, the long-term stability of CRISPR therapies remains uncertain, and off-target effects are a potential risk.
CRISPR gene editing technology
Another promising approach involves using CRISPR gene editing technology to reverse mutations in genes such as RNA binding motif protein 20 (RBM20), which are also associated with DCM. CRISPR gene editing has been shown to reverse mutations and cardiac dysfunction in mouse models of DCM. This technique targets specific genes, like RBM20, involved in heart function and holds promise for enhancing treatment outcomes for DCM patients. Therefore, CRISPR gene editing technology has the potential to be applied to a wide range of DCM-causing mutations, offering a broad therapeutic approach for this disease.
Stem cell therapy
Stem cell therapy involves transplanting stem cells into the heart to improve cardiac function. Research indicates that stem cell transplantation can improve left ventricular ejection fraction (LVEF), increase exercise capacity, and alleviate symptoms of heart failure in patients with DCM. Various stem cell types have been employed in DCM treatment, such as skeletal myoblasts, hematopoietic stem cells, mesenchymal stem cells, and CD34+ stem cells. Human pluripotent stem cell-derived cardiac cells are being explored for their potential in disease modeling, cell therapy, and drug discovery. However, stem cell therapy effectiveness remains uncertain due to limited high-quality studies and the need for further exploration.
Current therapeutic strategies for DCM are often limited in both effectiveness and accessibility, making heart transplantation the only viable option for patients with advanced stages of the disease. Many of these approaches rely on processes active in dividing cells, which limits their applicability for mature cells like cardiomyocytes and nerve cells that do not divide. However, recent advances in gene editing technologies like CRISPR-Cas9, stem cell therapy, gene modification, and others offer new, more effective, and targeted treatments that hold promise for improving treatment outcomes in DCM patients.

FAQs
In 2023, there were an estimated 554K diagnosed prevalent cases of DCM in the US, a figure expected to rise by 2034 due to a mix of genetic predispositions, lifestyle changes, and environmental factors. Importantly, about 40% of cases are hereditary, highlighting the need for genetic testing and precision approaches in management.
The DCM treatment market has long relied on symptom-management drugs such as ACE inhibitors, ARBs, beta-blockers, and diuretics. The only FDA-approved therapy specific to pediatric DCM is CORLANOR (ivabradine, Amgen), approved in 2019. While effective for heart rate control, it does not address the root genetic mechanisms of DCM, leaving a significant gap for disease-modifying therapies.
Standard care relies on ACE inhibitors, ARBs, beta-blockers, diuretics, and anticoagulants to manage symptoms and delay heart failure progression. Amgen’s CORLANOR (ivabradine) is the only FDA-approved therapy specifically for DCM, mainly prescribed for pediatric patients, though its use remains limited.
The DCM treatment market is driven by increasing disease prevalence, advances in genetic testing, and growing investment in regenerative and gene-based therapies. However, challenges remain: limited late-stage pipeline candidates, high costs of advanced treatments, and uncertainty around the long-term safety of novel modalities such as CRISPR and stem cells.
With gene therapy, stem cell interventions, and CRISPR editing gaining traction, the future DCM pipeline is shifting from symptom control toward actual disease modification. As clinical trials mature, it is anticipated that two-thirds of newly diagnosed DCM patients could see significant functional recovery within the first year of treatment, potentially transforming outcomes for this once-devastating condition.
Downloads
Article in PDF
Recent Articles
- Inozyme Raises $67M; Shanghai Miracogen partners with Synaffix; EdiGENE raises $15M; Antengene an...
- Takeda and AC Immune’s Alzheimer’s Deal; Eli Lilly’s Donanemab FDA Review; Bristol Myers Sq...
- Business Cocktail
- Lupin acquired; FDA grants approval; NATCO gets approval; Wockhardt Ltd. gets import alert; Paten...
- NgAgo gene-editing, claiming to be a better alternative to CRISPR gene editing, falls into a cont...



