Vertex & CRISPR Therapeutics Make History: FDA Approves exa-cel, the First CRISPR Gene Therapy for Sickle Cell and Beta-Thalassemia
Aug 22, 2025
Table of Contents
Vertex Pharmaceuticals and CRISPR Therapeutics have become the first companies to secure FDA approval for a CRISPR-based gene-editing therapy.
Vertex Pharmaceuticals, in partnership with CRISPR Therapeutics, has achieved a historic milestone with the FDA approval of exagamglogene autotemcel (exa-cel, branded as CASGEVY)—a first-ever CRISPR/Cas9–based gene therapy for sickle cell disease (SCD) and, later, transfusion-dependent beta-thalassemia (TDT). This landmark regulatory green light comes roughly seven years after exa-cel’s first clinical testing and ten years after CRISPR’s Nobel-winning discovery.
Downloads
Click Here To Get the Article in PDF

Recent Articles
- Aktis’s Novel Targeted Alpha Radiopharmaceuticals; Koye Partners with Sonde Health; Novartis to S...
- Gene Therapy to “Assassinate” HIV virus
- Oral Mucositis – A Common Cancer Therapy Adverse Reaction
- Key Clinical Outcomes- 30/07/2018
- Nucleic Acids and Gene Therapies in Neuromuscular Disorders: Next-Generation Therapeutic Strategies
From Discovery to FDA Approval
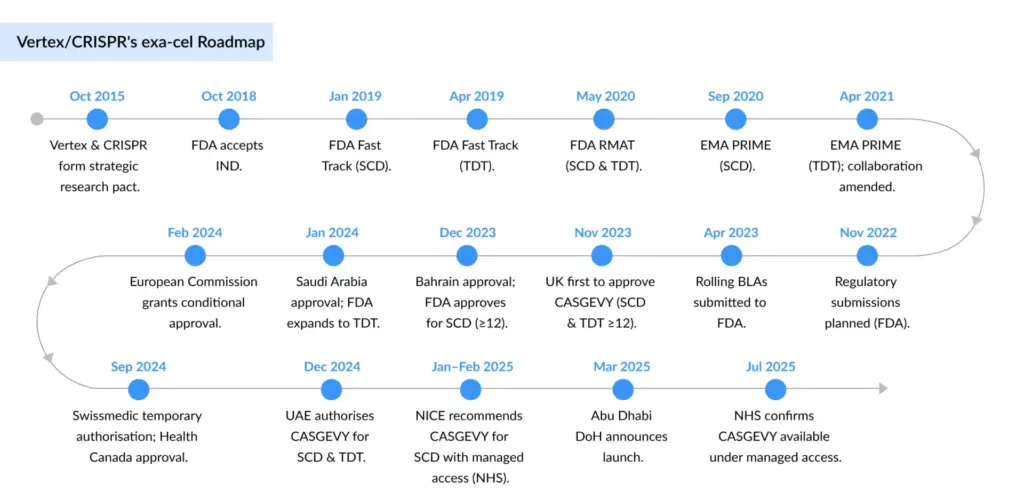
In 2015, Vertex and CRISPR Therapeutics entered a strategic research alliance to harness CRISPR/Cas9 for developing therapies that address the genetic roots of disease. Exa-cel is the first successful outcome of this collaboration. Under a revised partnership structure, Vertex leads global development, manufacturing, and commercialization, while program costs and profits are shared on a 60/40 basis with CRISPR Therapeutics.
In November 2022, Vertex and CRISPR Therapeutics initiated the final phase of regulatory engagement by submitting rolling Biologics License Applications (BLAs) to the FDA for exa-cel. The FDA accepted the submission in June 2023, granting Priority Review for the sickle cell disease (SCD) indication and Standard Review for transfusion-dependent beta-thalassemia (TDT), setting target action dates under the Prescription Drug User Fee Act (PDUFA): December 8, 2023, for SCD and March 30, 2024 for TDT.
By December 2023, the FDA granted full approval for exa-cel (marketed as CASGEVY) to treat patients aged 12 years and older with sickle cell disease and recurrent vaso-occlusive crises — making it the first CRISPR-based gene-editing therapeutic authorized in the US. The TDT indication followed shortly after, with FDA approval granted in January 2024. This milestone positions Exa-cel as Vertex’s most advanced program beyond its cystic fibrosis pipeline, solidifying its role as the pioneer in commercializing CRISPR-based gene editing therapies.
“Within a decade, we went from discovering the CRISPR platform to the first regulatory filings for a CRISPR-based therapy, demonstrating the transformative nature of CRISPR technology,” said Phuong Khanh (P.K.) Morrow, M.D., FACP, Chief Medical Officer at CRISPR Therapeutics.
Clinical Foundations and Evidence-Based
The FDA approval was anchored in robust clinical data from Phase III trials, notably CLIMB-121 and CLIMB-131, which demonstrated compelling results in SCD patients: among the 31 evaluable participants, 29 (93.5%) remained free of vaso-occlusive crises for at least 12 consecutive months, and all achieved freedom from hospitalization for VOCs during that period.
Vertex’s ongoing clinical program remains comprehensive, with additional trials—including CLIMB-111, CLIMB-141, CLIMB-151 (covering different age cohorts), and the long-term follow-up study CLIMB-131—continuing to generate data and monitor durability, safety, and long-term efficacy.
Exa-cel Mechanism of Action: How Does Exa-cel Work?
Exa-cel is an autologous, ex vivo CRISPR/Cas9 gene-edited therapy. Patients undergo mobilization and harvesting of their hematopoietic stem and progenitor cells, which are then gene-edited to reactivate fetal hemoglobin (HbF) production in red blood cells. Following myeloablative conditioning, the edited cells (exa-cel) are infused back into the patient, engraft in the bone marrow, and begin producing healthier hemoglobin, effectively alleviating disease manifestations. The side effect profile is consistent with autologous stem cell transplantation and includes cytopenias, mucositis, nausea, musculoskeletal pain, febrile neutropenia, and other manageable effects.
“As we move into the second half of the year, we are building strong momentum across both our commercial and clinical programs,” said Samarth Kulkarni, Ph.D., Chairman and CEO of CRISPR Therapeutics. “With 75 authorized treatment centers for CASGEVY now activated, we have taken a significant step forward in broadening patient access.”
Regulatory Momentum and Global Reach
Exa-cel’s trajectory was accelerated by a suite of US FDA designations, Regenerative Medicine Advanced Therapy (RMAT), Fast Track, Orphan Drug, and Rare Pediatric Disease — which not only shortened review timelines but highlighted its transformative potential.
Regulatory progress in Europe and the UK has been equally intense. Marketing Authorization Applications (MAAs) were submitted in late 2022 and validated in early 2023 by both the European Medicines Agency (EMA) and the UK Medicines and Healthcare Products Regulatory Agency (MHRA). Exa-cel went on to secure Orphan Drug Designation from the European Commission, PRIME status from the EMA, and an Innovation Passport under the ILAP pathway from the MHRA — each underscoring broad regulatory alignment on its potential to shift standards of care.
“Even if access were opened today, I’d expect no more than about 10% of sickle cell patients to pursue this therapy. And even that level of demand would exceed our current capacity to manage.”
Dr. Julie Kanter – Director, Adult Sickle Clinic, UAB; President, National Alliance of Sickle Cell Centers
Regulatory Oversight and Long-Term Safety
The FDA requires patients treated with gene therapy to be monitored for up to 15 years for potential late-onset adverse events. For CASGEVY, this mandate translates into two post-marketing requirement (PMR) safety studies, focused on assessing risks of hematologic malignancies and unintended off-target genome edits. Both studies align with the FDA’s industry-wide guidance on long-term follow-up after administration of human gene therapies.
Beyond clinical surveillance, CASGEVY is protected by basic product patents until 2035 in the US and 2034 in Europe, providing a strong intellectual property barrier to competition during its early commercialization years.
Reimbursement and Access Expansion
On the access side, CASGEVY is already broadly reimbursed across US third-party payors, including federal programs. Currently, all eligible patients are covered through single-case agreements, with national and regional payor contracts collectively spanning more than 250 million lives. Importantly, CASGEVY secured a new technology add-on payment for Medicare SCD patients, and reimbursement pathways have been confirmed with Medicaid in all 25 priority states where SCD prevalence is highest. In parallel, Vertex is participating in the Cell and Gene Therapy (CGT) Access Model, a novel CMS-led initiative designed to accelerate broad Medicaid coverage through outcomes-based agreements. Internationally, reimbursement discussions are underway, with England granting national reimbursement and early access programs anticipated in additional markets.
Manufacturing, Pricing, and the Economics of Cure
Vertex and CRISPR have already achieved their target of 75 authorized treatment centers (ATCs) worldwide, marking a pivotal milestone in the commercial rollout. Momentum is accelerating: as of June 30, 2025, more than 115 patients have completed initial cell collection, and 29 have received CASGEVY infusions — including 16 in the second quarter alone. These early adoption figures underscore both growing demand and the scalability of the program..
Pricing remains a defining challenge. Severe SCD patients in the US often incur lifetime costs of USD 1.5–2 million in hospitalizations, transfusions, and complications. Against this backdrop, a one-time curative therapy is potentially cost-saving despite a steep upfront price. Exa-cel has been set at around USD 2.2 million per patient — placing it among the most expensive therapies ever launched, but still within the bounds of current gene therapy economics. Innovative payment models, including installment-based and outcomes-linked contracts, are expected to play a key role in driving adoption.
“These therapies finally offer options for conditions where treatment has long been unsatisfying. But the immediate reality is that the price is extraordinarily high, and state budgets simply cannot shoulder that alone.”
Kate McEvoy – Executive Director, National Association of Medicaid Directors
Competitive Landscape and Payer Strategy
Pricing pressures will be compounded by competition. Bluebird Bio is positioning lovo-cel for SCD and ZYNTEGLO for TDT, both of which rely on viral vector gene addition. ZYNTEGLO already carries a USD 2.8 million price tag, and Bluebird’s struggle to secure reimbursement in Europe — ultimately prompting its market exit — underscores the hurdles Vertex and CRISPR must navigate.
Learning from that precedent, Vertex has adopted an aggressive payer engagement strategy. The company has already met with all US state Medicaid agencies, ~150 commercial payers, and leading European health technology assessment bodies, including NICE in the UK. According to COO of Vertex, Stuart Arbuckle, payers have described exa-cel’s clinical data as “highly impactful,” particularly in reducing hospitalizations and vaso-occlusive crises.
Critically, exa-cel’s mechanistic differentiation sets it apart from Bluebird’s therapies. Instead of inserting a working copy of the HBB gene, exa-cel uses CRISPR/Cas9 to switch on fetal hemoglobin production, restoring oxygen delivery and reducing anemia. Vertex estimates that ~32,000 patients in the US could be eligible, highlighting both the scale of the opportunity and the logistical challenges of delivering such a complex therapy.
The Road Ahead: Beyond a Scientific First
CASGEVY is not just a new medicine; it is a proof point for the viability of gene editing as a therapeutic modality. Yet, its success will depend as much on economics and infrastructure as on science. Ensuring equitable access, securing reimbursement at scale, and building durable long-term safety datasets are the next tests for Vertex and CRISPR.
If they succeed, CASGEVY could redefine what a “cure” looks like in hematology and open the door for a wave of CRISPR-based treatments across genetic diseases. The approval marks the beginning of a new chapter — one where the promise of rewriting DNA is no longer aspirational but a clinical reality.
FAQs
Sickle cell disease is an inherited blood disorder caused by mutations in the gene responsible for hemoglobin production. Instead of flexible, round red blood cells, patients produce rigid, sickle-shaped cells that break down prematurely. This not only leads to chronic anemia but also obstructs blood vessels, driving pain crises, organ damage, and reduced life expectancy.
Symptoms typically emerge in early infancy, often around six months of age. While severity varies widely, hallmark complications include chronic anemia, debilitating pain episodes, swelling in the hands and feet, recurrent infections, and progressive vision impairment. Over time, these manifestations can significantly compromise quality of life and increase healthcare dependence.
The only established cure remains allogeneic stem cell transplantation, though it is feasible for only a small fraction of patients due to donor limitations and procedural risks. For most, management involves hydroxyurea, transfusions, pain control, and newer disease-modifying therapies that reduce complications. The emergence of gene-editing therapies such as CASGEVY represents a paradigm shift, aiming to address the root cause rather than just symptoms.
Beta-thalassemia is a genetic blood disorder driven by mutations in the beta-globin gene, leading to insufficient hemoglobin and defective red blood cell production. This chronic shortage of healthy red cells results in lifelong anemia and drives complications ranging from bone deformities and organ overload to growth delays.
Management depends on disease severity. Patients with transfusion-dependent beta-thalassemia (TDT) require regular lifelong transfusions and iron chelation therapy, while non–transfusion-dependent patients (NTDT) may still need intermittent interventions. Despite advances in supportive care, the treatment burden remains high, underscoring the need for transformative options such as gene therapies that could potentially replace decades of transfusion dependency.

Downloads
Article in PDF



