Will The Burgeoning Gene Therapies Make a Difference in Dystrophic Epidermolysis Bullosa Patients’ Lives?
Jun 16, 2023
Table of Contents
Dystrophic epidermolysis bullosa (DEB) is an inherited disorder caused by changes in the COL7A1 gene. In this rare condition, blisters form on the skin and the moist inner lining of some organs and body cavities. Depending upon the nature of the inheritance pattern, dystrophic epidermolysis bullosa is divided into recessive dystrophic epidermolysis bullosa (RDEB) and dominant dystrophic epidermolysis bullosa (DDEB). In the United States, around 3,500 people were diagnosed with dystrophic epidermolysis bullosa in 2021, as per DelveInsight. Out of these, approximately 1,100 people were severely affected.
The current dystrophic epidermolysis bullosa treatment goal is to manage blisters and erosions, control infection, and prevent complications. Besides, symptomatic relief is also taken care of as pain and itch severely harm the patient’s quality of life. Moreover, surgical dystrophic epidermolysis bullosa treatments provide a symptomatic improvement from many deformities and skin tumors.
Downloads
Click Here To Get the Article in PDF

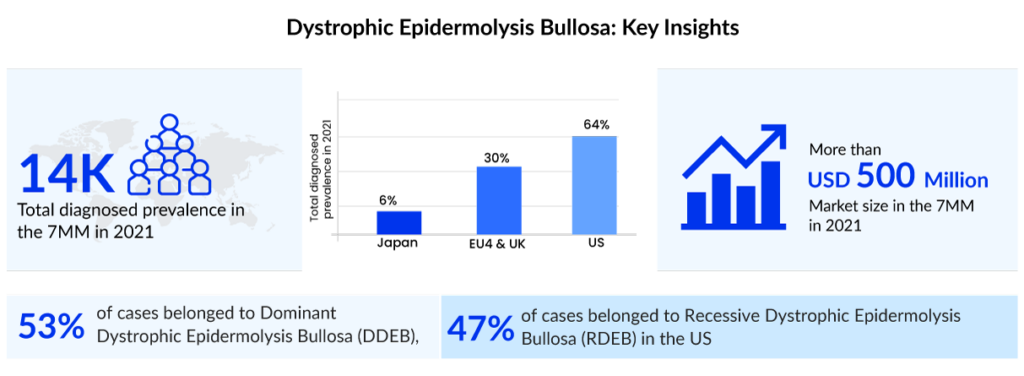
According to DelveInsight’s estimates, the current symptomatic market of dystrophic epidermolysis bullosa is around USD 350 million in the United States. Advancements in genetic testing and disease understanding are likely to fuel the dystrophic epidermolysis bullosa treatment market growth. The respective portfolios of several biopharma companies feature multiple pipeline projects focused on the development of newer treatment options like cell-based therapies, gene therapies, protein replacement therapies, and autologous and allogeneic stem cell therapies, among others. Gene therapies, which involve the transfer of functional COL7A1 gene to the patient with RDEB, appear to be at the forefront as promising potential treatment options.
Entry of VYJUVEK: First-ever redosable gene therapy
Recently, on 19 May 2023, Krystal Biotech, Inc., a biotechnology company focused on developing and commercializing genetic medicines for patients with rare diseases, announced that the FDA has approved VYJUVEK (beremagene geperpavec-svdt) for the treatment of dystrophic epidermolysis bullosa patients 6 months of age or older. VYJUVEK is intended to treat the genetic root cause of DEB by delivering functional copies of the human COL7A1 gene, resulting in wound healing and sustained functional COL7 protein expression with redosing. VYJUVEK is the first and only FDA-approved drug for dystrophic epidermolysis bullosa treatment, both recessive and dominant, that can be administered by a healthcare professional in either a healthcare professional setting or at home.
VYJUVEK was approved by the FDA based on two clinical investigations. The GEM-1/II trial was an intra-patient, open-label, single-center, randomized, placebo-controlled study that found that repeat topical applications of VYJUVEK were associated with long-term wound closure, full-length cutaneous COL7 expression, and anchoring fibril assembly with few adverse events. The GEM-3 experiment was an intra-patient, double-blind, multicenter, randomized, placebo-controlled study that fulfilled both its primary and secondary endpoints of full wound healing at six months and three months. VYJUVEK was well tolerated, with no drug-related major adverse events or treatment-related discontinuations.
VYJUVEK will be accessible in the United States in the third quarter of 2023, and the Company will begin marketing it immediately. Krystal Connect, a personalized support program, was designed by the Company to fulfill the needs of patients, carers, and families as they begin and continue their VYJUVEK treatment journey. The program comprises services that may answer VYJUVEK-related queries, verify health benefits, aid with treatment planning and administration, and provide information about financial assistance for qualified patients. Patients, carers, and healthcare professionals can get further information by calling 1-844-5-KRYSTAL.
The FDA granted the company a Rare Paediatric Disease Priority Review Voucher (PRV), which grants priority review to a subsequent medication application that would not otherwise qualify for priority review. The PRV program promotes the discovery of novel medications for the prevention or treatment of rare diseases.
Outside of the United States, the European Medicines Agency has designated VYJUVEK as an orphan drug and granted it PRIME (PRIority MEdicines) status for the treatment of dystrophic epidermolysis bullosa. The Company expects to begin the official Marketing Authorization Application procedure in the second half of 2023, with approval possible in 2024. The company is also collaborating with Japan’s Pharmaceuticals and Medical Devices Agency to examine VYJUVEK and seek permission for a possible launch in 2025.
Potential pipeline assets for dystrophic epidermolysis bullosa treatment
Many companies are developing possible treatments for dystrophic epidermolysis bullosa with continuous efforts in research and development. Castle Creek Biosciences (FCX-007), Amryt Pharma (Oleogel-S10), Abeona Therapeutics (EB-101), and Phoenix Tissue Repair (PTR-01) are key players working on potential dystrophic epidermolysis bullosa therapies like gene therapy, symptomatic relief therapy, and disease-modifying therapies.

FCX-007 (Dabocemagene autoficel) is an investigational gene therapy to address functional type VII collagen protein (COL7) deficiency in patients with DEB. It is a genetically-modified autologous fibroblast that encodes the gene for COL7 and treats wounds locally via injection. Further, EB-101 an autologous, gene-corrected cell therapy for RDEB involves using gene transfer technology to deliver COL7A1 genes into an RDEB patient’s skin cells (keratinocytes) and transplanting them back to the patient to enable normal type VII collagen expression.
Apart from the promising gene therapies, other therapies are also in the dystrophic epidermolysis bullosa pipeline. PTR-01 is an intravenous protein replacement therapy that uses a recombinant collagen type VII (Rc7) to treat RDEB. Apart from this, RGN-137 (Lenus Therapeutics) and INM-755 (InMed Pharmaceutical) are the two topical therapies recruiting subjects in the Phase II trial of clinical development for treating Epidermolysis Bullosa.
By successfully testing and launching these emerging therapies, the total dystrophic epidermolysis bullosa treatment market will increase and is poised to reach approximately USD 1 billion in the seven major markets by 2032.

Clinical challenges remain
With the advent of curative therapies, the upcoming dystrophic epidermolysis bullosa therapeutic landscape looks promising. However, a few challenges need to be looked at; ex-vivo autologous gene replacement therapy involves isolating patient cells, genetically correcting them in vitro by introducing wild-type cDNA copies of the affected gene, expanding the corrected cells into epidermal sheets, and grafting these back onto chronic wounds. This form of therapy is mainly investigated in severe EB types (JEB, RDEB), which typically display an autosomal recessive inheritance.
Moreover, the current gene delivery techniques cannot specifically target epidermal stem cells called holoclones in vivo. Further, ensuring efficient targeting of these epidermal stem cells first requires their isolation before introducing the replacement gene ex-vivo. However, proper signaling is restored upon reintroducing the wild-type LAMB3 cDNA, thereby imparting a replicative advantage to corrected clones in JEB.
However, this is not the case with RDEB, which poses several challenges, like the significantly greater size of the COL7A1 transcript, the YAP/TAZ pathway being unaffected, and transduced clones not gaining a replicative advantage. Consequently, this approach is far less prominent in RDEB. Besides, gene-targeted therapies face common issues of efficacy, safety, feasibility, tolerability, and cost-effectiveness.
Will payers embrace the wave of costly gene therapies?
Reimbursement of durable and potentially curative dystrophic epidermolysis bullosa gene therapies will challenge payers in US and EU regions due to their high upfront costs. The lack of long-term clinical studies magnifies uncertainties which will be challenging for payer coverage and reimbursement decisions. These characteristics could negatively impact patient access and, ultimately, future developer innovation.

FAQs
Dystrophic epidermolysis bullosa is one of the most common types of epidermolysis bullosa. Epidermolysis bullosa causes the skin to be extremely delicate and to blister readily.
The common signs and dystrophic epidermolysis bullosa symptoms include blisters, itchy, painful skin, dysphagia, dental problems, and others.
The dystrophic epidermolysis bullosa causes are generally inherited. The disease’s gene may be passed down from one of the disease’s parents (autosomal dominant inheritance)
The current dystrophic epidermolysis bullosa treatment includes contemporary wound care and the reduction of external factors that cause blistering and hinder wound healing. A high degree of personal hygiene and intense moisturizing dystrophic epidermolysis bullosa treatment is required for proper skin care.
Downloads
Article in PDF
Recent Articles
- Plasma Proteins Therapeutics: Transforming Treatment in Hematology and Beyond
- WASKYRA Wins FDA Approval, Marking a Historic Leap for Rare Disease Gene Therapies
- Rare Disease Day
- Watershed Moment for Cell Therapies and Complicated Journey of Gene Therapies in Japan
- FDA Approves Sarepta’s Muscular Dystrophy Drug after Months of Debate



