Another Feather in the Cap for Xtandi and Keytruda — The Two Main Cancer Drugs
Nov 27, 2023
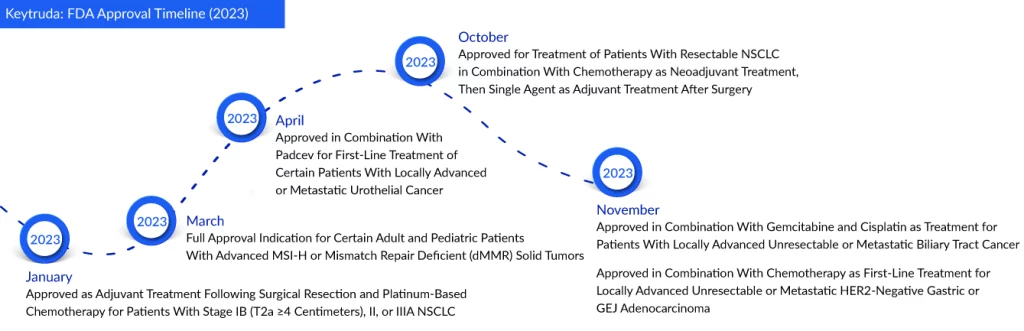
The FDA has approved label extensions for two of the most crucial cancer medications globally—Merck’s Keytruda and Pfizer and Astellas’ Xtandi. Keytruda’s expanded indication now includes stomach cancer, permitting its usage alongside chemotherapy for first-line treatment in patients with locally advanced unresectable or metastatic HER2-negative gastric or gastroesophageal junction (GEJ) adenocarcinoma.
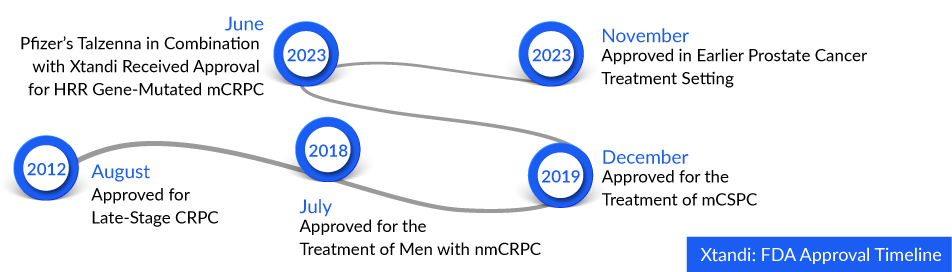
Xtandi’s expanded approval broadens its comprehensive portfolio in prostate cancer treatment. It is now the sole androgen receptor inhibitor sanctioned by the FDA for patients with nonmetastatic castration-sensitive prostate cancer (nmCSPC) experiencing biochemical recurrence at high risk for metastasis (high-risk BCR). Treatment options include Xtandi, either alone or in combination with GnRH analog therapy. Statistics indicate that 20% to 40% of men who undergo prostate cancer treatment will encounter BCR within a decade. Moreover, among those with high-risk BCR, 90% are expected to progress to metastatic disease, with one in three facing mortality.

“For individuals who had successfully undergone treatment for prostate cancer and experienced a period of remission, the news of disease recurrence and the looming risk of metastasis can be emotionally overwhelming,” noted Courtney Bugler, President and CEO of ZERO Prostate Cancer. “The approval of XTANDI represents a hopeful treatment avenue for the community, providing a glimmer of hope to patients and their caregivers in the face of these difficult circumstances.”
Downloads
Click Here To Get the Article in PDF

Recent Articles
- Telegenomics nets $23M; Merck acquires Tilos; ADC nabs; Sobi buys Novoimmune’s assets
- Merck’s Keytruda sales; Valeant on name change; Pfizer – BMS; Amgen puts Repatha outcomes for deal
- Gilead’s Magrolimab Plus Azacitidine for MDS; FDA Approveds VANFLYTA for Newly Diagnosed AML; FDA...
- Copiktra receives approval; Lilly wins approval; Eisai’s Fycompa; Astellas’ Roxadustat; Janssen’s...
- Takeda’s ADZYNMA Approved by FDA; AskBio Presents Preliminary Data from Phase I Trial of Gene The...
The green light for this approval is grounded in the outcomes of the Phase III EMBARK trial. This trial investigated the effectiveness of XTANDI in conjunction with leuprolide, a placebo paired with leuprolide, and XTANDI as a standalone therapy in patients grappling with nonmetastatic hormone- (or castration-) sensitive prostate cancer (nmHSPC or nmCSPC) featuring high-risk biochemical recurrence (BCR). Detailed trial results took center stage during a plenary session at the 2023 American Urological Association Annual Meeting and were subsequently published in the New England Journal of Medicine.
“After more than a decade of dedicated research and development, this FDA approval marks a significant achievement for us. We’ve been committed to advancing XTANDI for the benefit of as many prostate cancer patients as possible,” said Dr. Ahsan Arozullah, Senior Vice President and Head of Oncology Development at Astellas. “Our clinical development program has been pivotal in transforming the lives of patients, and we’re particularly proud that XTANDI is now available for a specific group of men with nonmetastatic castration-sensitive prostate cancer experiencing biochemical recurrence and high metastatic risk.”
“The approval to expand XTANDI’s indication into an earlier stage of the disease is a significant milestone, especially considering that more than 300,000 men in the U.S. have already been prescribed this medication,” stated Dr. Chris Boshoff, Chief Oncology Research and Development Officer and Executive Vice President at Pfizer. “XTANDI has established a robust clinical profile, demonstrating overall survival benefits for patients with metastatic castration-resistant prostate cancer, nonmetastatic castration-resistant prostate cancer, and metastatic castration-sensitive prostate cancer. This approval opens doors to providing this therapy to a broader group of patients with nonmetastatic castration-sensitive prostate cancer at high risk.”
Xtandi holds the distinction of being the exclusive novel hormone therapy given the green light for treating three distinct prostate cancer types. The sales of Xtandi experienced a substantial uptick in 2022, reaching $5.9 billion—a remarkable 27% surge compared to the previous year. XTANDI is presently in the review process with international regulatory authorities, such as the European Medicines Agency, as part of efforts to secure an expanded indication for nmHSPC (or nmCSPC) with high-risk BCR, supported by the robust results emanating from the EMBARK study.
Regarding Keytruda, it has received its seventh approval for use in gastrointestinal cancers in the United States, marking its 38th overall indication as a checkpoint inhibitor. Previously, Keytruda was designated for gastric cancer only in patients who showed progression following a minimum of two prior courses of systemic therapy, which incorporated fluoropyrimidine- and platinum-based chemotherapy. This year, it is expected to surpass AbbVie’s Humira as the world’s best-selling drug. Merck recorded a sales figure of $20.9 billion for Keytruda last year, representing a 22% growth from 2021.
Dr. Marjorie Green, senior vice president and head of late-stage oncology, global clinical development at Merck Research Laboratories, shared, “Our extensive development program at Merck spans a wide spectrum of gastrointestinal cancers, aiming to introduce meaningful new choices for patients and their healthcare providers. The recent approval of a treatment option based on KEYTRUDA marks a significant achievement for individuals with advanced HER2-negative gastric or GEJ adenocarcinoma. This milestone underscores Merck’s unwavering dedication to addressing the specific needs of these patients in the United States.”
The FDA granted approval based on findings from the Phase III KEYNOTE-859 study. This randomized, double-blinded trial involved 1,579 participants who had not undergone systemic therapies. Results revealed a 22% reduction in the risk of death with the Keytruda-based regimen compared to chemotherapy alone. Additionally, median survival was marginally extended in the Keytruda arm.
Merck’s flagship in oncology, Keytruda, is a monoclonal antibody strategically designed to target the PD-1 receptor. Its mechanism involves disrupting the downstream signaling of PD-1, leading to a heightened immune response against cancer cells. Remarkably, Keytruda has been deployed in more than 1,600 trials across diverse cancer types and treatment modalities.
Recently, Merck also obtained approval for Keytruda’s expanded use in biliary tract cancer, where it is now incorporated into a combination regimen with gemcitabine and cisplatin chemotherapy. The FDA granted approval following the analysis of results from Merck’s Phase III KEYNOTE-966 trial, revealing a noteworthy increase in overall survival when the drug was administered alongside chemotherapy, as opposed to chemotherapy alone.
Additionally, on 10 November 2023, Europe’s Committee for Medicinal Products for Human Use also recommended the approval of Keytruda for use alongside the antimetabolite gemcitabine and chemotherapy. This recommendation is for the primary treatment of adults diagnosed with locally advanced unresectable or metastatic biliary tract cancer.
Keytruda is facing heightened competition in the biliary tract cancer treatment sector, with AstraZeneca’s Imfinzi gaining FDA approval in September of last year, particularly in combination with chemotherapy. AstraZeneca underscores a notable 20% risk reduction in mortality based on its Phase III trial, and the drug has propelled AstraZeneca’s earnings, contributing over $1.9 billion in the first half of 2023.
Despite this, Keytruda has further solidified its status as one of Merck’s main revenue generators, securing another FDA approval. In the third quarter of 2023 alone, Keytruda recorded sales exceeding $6.3 billion, marking a 17% surge compared to the corresponding period last year. Cumulatively for this year, the drug has amassed sales surpassing $18.4 billion.
In October 2023, Merck achieved a milestone by extending the application of Keytruda to the perioperative phase for non-small cell lung cancer treatment. The FDA’s approval now permits the use of the PD-1 blocker as a neoadjuvant treatment in conjunction with platinum-based chemotherapy, followed by its use as an adjuvant monotherapy post-surgery.
Moreover, the recent approval for Keytruda in GEJ adenocarcinoma is once again intensifying the competition with its counterpart, Opdivo from Bristol Myers Squibb. Opdivo received the first immunotherapy approval for stomach cancer in 2021, making the rivalry even more significant. Agilent Technologies also gained FDA approval recently for its diagnostic assay, enabling the identification of gastric and gastroesophageal junction adenocarcinoma patients suitable for Keytruda’s newly approved combination therapy.

Downloads
Article in PDF
Recent Articles
- ADCs in Lung Cancer Treatment: ENHERTU’s Rise, HER3 & TROP-2 Challenges, and What’s Next in ...
- Human Papillomavirus (HPV) Prophylaxis– Expected to see a steady growth from 2015-2025
- Teva’s AJOVY Expanded by FDA as First Anti-CGRP Preventive for Pediatric Episodic Migraine; NRx P...
- Phase III RUBY Trial of Jemperli Plus Chemotherapy Updates; FDA Approves Roche’s Vabysmo for RVO;...
- Astellas & AviadoBio’s Exclusive Deal for AVB-101; GSK’s Depemokimab Shows Positive Res...



