Top FDA Approvals to Watch in H2 2026: Regulatory Decisions Poised to Transform the Pharmaceutical Landscape
Jul 17, 2026
Table of Contents
Summary
- H2 2026 is expected to be a highly active FDA regulatory period, with decisions spanning oncology, nephrology, infectious diseases, neurology, hematology, and rare diseases, reflecting greater therapeutic diversity beyond oncology dominance.
- Key innovation trends include first- and best-in-class biologics, RNA-based therapies, long-acting antivirals, precision oncology drugs, advanced imaging diagnostics, and patient-friendly subcutaneous formulations.
- Promising candidates that are expected to launch in H2 2026 include Celcuity’s gedatolisib, Replimune’s vusolimogene oderparepvec (RP1), Merck’s subcutaneous KEYTRUDA, Gilead Sciences’ bictegravir plus lenacapavir, Lantheus’ MK-6240, Nuvalent’s zidesamtinib, Novo Nordisk’s denecimig (Mim8), Savara’s MOLBREEVI, and BioMarin’s BMN 351.
The second half of 2026 is set to be one of the busiest regulatory periods for the biopharmaceutical industry, with the US FDA expected to announce decisions on several high-profile therapies across oncology, nephrology, infectious diseases, neurology, hematology, and rare diseases. Unlike previous years, when oncology dominated the regulatory calendar, H2 2026 reflects increasing therapeutic diversity, featuring first- and best-in-class biologics, targeted therapies, RNA-based medicines, long-acting antivirals, advanced diagnostic imaging agents, and patient-friendly subcutaneous formulations of established blockbuster therapies. Collectively, these anticipated approvals underscore the industry’s continued shift toward precision medicine, improved patient convenience, and innovative therapies addressing high unmet medical needs.
Downloads
Click Here To Get the Article in PDF

July Opens with Major Opportunities in Nephrology and Oncology
The H2 2026 regulatory calendar begins with a major milestone in nephrology. Vera Therapeutics’ TRUTAKNA (atacicept) received FDA accelerated approval for IgA nephropathy (IgAN) based on positive Phase III ORIGIN 3 results demonstrating a 46% reduction in proteinuria. As the first dual BAFF/APRIL inhibitor, TRUTAKNA targets the underlying immunologic drivers of IgAN with a price annualizing to more than USD 400,000 per patient. The once-weekly subcutaneous therapy is expected to compete with approved and late-stage candidates, including VOYXACT (Otsuka), Ultomiris (AstraZeneca), Sefaxersen (Ionis Pharmaceuticals), Felzartamab (Biogen), Sparsentan (Chugai/Renalys), and BHV-1400 (Biohaven).
Industry Expert’s sentiments: While some nephrologists have high enthusiasm for TRUTAKNA due to its ability to replace steroid use and address the root cause, others note that its weekly dosing may be a disadvantage compared to once-monthly competitors like VOYXACT and povetacicept. As far as Market adoption is concerned, KOLs anticipate a gradual adoption curve as community nephrologists become more comfortable with these new biologics.
According to Sadaf Javed, Functional Head of Forecasting & Analytics at DelveInsight, TRUTAKNA is projected to generate approximately USD 1.6 billion in market revenue by 2036, while continued approval remains contingent upon confirmatory evidence of kidney function preservation.
Also in July, Celcuity’s gedatolisib is under FDA Priority Review for the treatment of HR-positive, HER2-negative, PIK3CA wild-type advanced breast cancer, with a PDUFA date of July 17, 2026. Supported by positive Phase III VIKTORIA-1 data, the NDA is being reviewed under the FDA’s Real-Time Oncology Review (RTOR) program. The dual PI3K/mTOR inhibitor has also received Breakthrough Therapy and Fast Track designations for patients progressing after CDK4/6 inhibitor therapy. If approved, gedatolisib could become the first targeted therapy for the PIK3CA wild-type population, targeting roughly 40,000 patients in the US who receive 2nd line treatment post CDK4/6 inhibitors, strengthening Celcuity’s position in the competitive HR+/HER2− breast cancer market.
August Features Multiple High-Impact Regulatory Catalysts
August is expected to be one of the busiest months on the H2 2026 FDA calendar, with several high-profile regulatory decisions spanning oncology, infectious diseases, and neurodegenerative disorders.
Replimune’s vusolimogene oderparepvec (RP1), in combination with nivolumab, is under FDA review for advanced melanoma, with a PDUFA date of August 2, 2026. The resubmitted BLA, supported by updated Phase II IGNYTE data, demonstrated a median overall survival of 32.9 months, an objective response rate of 33.6%, and a median duration of response of 24.8 months. If approved, RP1 could offer a novel treatment option for patients with checkpoint inhibitor-resistant melanoma and further validate oncolytic virotherapy in immuno-oncology. The therapy is expected to compete with emerging candidates, including Brenetafusp (Immunocore; Phase III), iSCIB1+ (Scancell Holdings; Phase II), and Intismeran (Moderna; Phase II), further intensifying competition in the evolving melanoma treatment landscape.
Industry Expert’s sentiments: Industry experts express a mix of optimism regarding efficacy and caution regarding real-world adoption. While some oncologists were “impressed” by the IGNYTE trial data, noting it as a more tolerable and efficacious combination for post-PD-1 melanoma, others are concerned about significant administration (intratumoral) hurdles impacting long-term performance.
Merck’s subcutaneous KEYTRUDA (pembrolizumab) is among the most anticipated regulatory decisions of H2 2026. The FDA has granted Priority Review to the supplemental BLA for KEYTRUDA QLEX in combination with Padcev for muscle-invasive bladder cancer (MIBC), with a PDUFA date of August 17, 2026. If approved, the subcutaneous formulation could improve patient convenience, reduce infusion chair time, and optimize healthcare resource utilization while maintaining efficacy. The approval would further strengthen KEYTRUDA’s market leadership, as Merck aims to transition 30-40% of the KEYTRUDA market to the SubQ version.
Industry Expert’s sentiments: In the MIBC setting, experts anticipate that the SubQ formulation will become a mainstay as a maintenance therapy, and it is also going to save some time for both physicians and patients as IV administration typically requires a 30 to 60-minute infusion, whereas the SubQ injection is completed in just one to three minutes, resulting in improvement in compliance.
Despite the operational advantages, experts highlight several hurdles that may slow the total conversion from IV to SubQ, due to IV biosimilar entry and reimbursement challenges
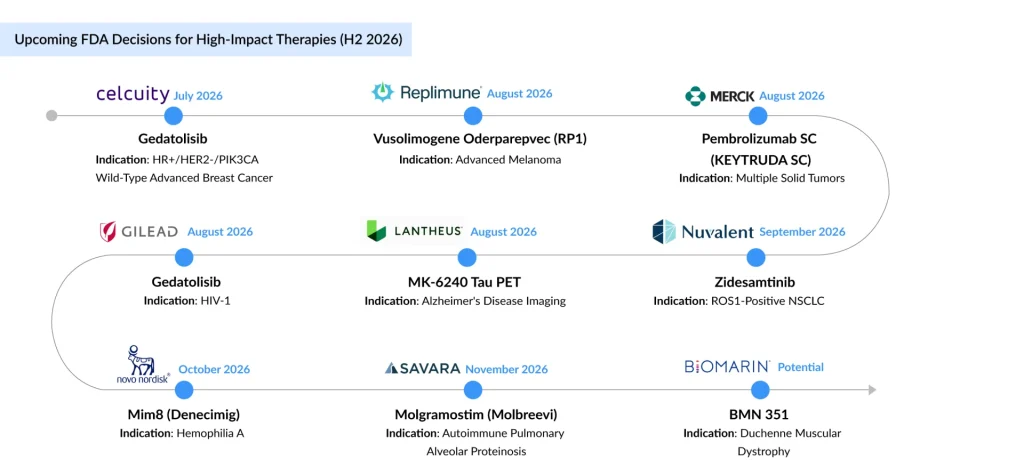
Gilead Sciences’ bictegravir plus lenacapavir (BIC/LEN) is under FDA Priority Review, with a decision expected on August 27, 2026. Designed as a once-daily single-tablet regimen for virologically suppressed adults with HIV-1, BIC/LEN combines bictegravir’s high barrier to resistance with the novel capsid inhibitor lenacapavir. If approved, it would become the smallest single-tablet HIV regimen, offering a simplified maintenance option to improve long-term adherence. The therapy will compete with ViiV Healthcare’s VH184, VH499, and ultra-long-acting cabotegravir (all Phase II), while Merck and Gilead continue to advance the once-weekly oral islatravir plus lenacapavir regimen in the Phase III ISLEND-1 and ISLEND-2 trials, further intensifying competition in the evolving HIV treatment landscape.
Industry Expert’s sentiments: Gilead’s strength lies in its ability to combine complementary mechanisms into easy-to-administer treatments. There is a strong industry perception that Gilead’s longevity in the HIV market equates to superior medicine.
Lantheus’ MK-6240 is under FDA review as an ^18F-labeled tau PET imaging agent for patients undergoing evaluation for Alzheimer’s disease, with a PDUFA date of August 13, 2026. Backed by two pivotal Phase III studies demonstrating high sensitivity and specificity for detecting tau neurofibrillary tangles, MK-6240 has the potential to improve diagnostic accuracy and support earlier identification of patients eligible for disease-modifying therapies. The imaging agent is expected to compete with GE HealthCare’s Vizamyl (flutemetamol F 18), approved for beta-amyloid PET imaging, and Abbott’s blood-based pTau217 assay, developed in partnership with ALZpath , as precision diagnostics continue to transform Alzheimer’s disease management.
Industry Expert sentiments: Experts believe amyloid imaging will serve as the front-line for early identification, while tau PET agents like MK-6240 will provide the granularity needed for longitudinal monitoring. However, there are potential headwinds, including the rise of blood biomarker testing, which could eventually decrease the overall demand for PET imaging if biomarkers prove sufficiently accurate for routine screening.
Precision Medicine Dominates September
September continues the momentum with a key regulatory decision in precision oncology. Nuvalent’s zidesamtinib is under FDA review for TKI-pretreated ROS1-positive advanced non-small cell lung cancer (NSCLC), with a PDUFA date of September 18, 2026. Supported by Phase I/II ARROS-1 data, the therapy demonstrated a 44% objective response rate and durable responses, with 6-, 12-, and 18-month duration of response rates of 84%, 78%, and 62%, respectively. Designed to overcome acquired ROS1 resistance mutations while maintaining robust central nervous system (CNS) activity, zidesamtinib addresses a key unmet need after prior ROS1 TKI therapy. If approved, zidesamtinib is expected to compete with approved ROS1 inhibitors, including IBTROZI/DOVBLERON (Innovent Biologics) and AUGTYRO (Bristol Myers Squibb/Zai Lab), as well as late-stage candidates from AnHeart Therapeutics, Eisai, and Merck Sharp & Dohme’s sacituzumab tirumotecan (Phase III).
Industry Expert Sentiments: Experts view zidesamtinib as a best-in-class asset with significant revenue potential due to its ability to drive durable responses and capture market share from earlier-generation therapies.
As per Javed, zidesamtinib is projected to generate approximately USD 400 million in market revenue by 2036, reinforcing its potential to become a significant player in the evolving ROS1-positive NSCLC treatment landscape.
October Highlights Rare Diseases Innovation
October is expected to feature a key regulatory decision in hematology, with Novo Nordisk’s denecimig (Mim8) emerging as one of the most closely watched therapies for hemophilia A.
Denecimig (Mim8) is an investigational FVIIIa-mimetic bispecific antibody for prophylactic treatment of hemophilia A, with or without inhibitors. Supported by data from the comprehensive FRONTIER clinical program, the therapy demonstrated favorable efficacy, safety, and flexible dosing across multiple patient populations. An FDA decision is anticipated in October 2026 following BLA submission, positioning denecimig as a potential competitor to Hemlibra. If approved, it will enter an increasingly competitive market alongside recently approved therapies, including HYMPAVZI (Pfizer), ALHEMO (Novo Nordisk), and QFITLIA (Sanofi), as well as NXT007 (Roche/Chugai), currently in Phase III development.
Industry Expert Sentiments: Some hematologists see meaningful advantages in Mim8’s trial data that could drive adoption, particularly for patients seeking improved bleed control. Whereas other experts predict very little interest in switching for patients who are already well-controlled on HEMLIBRA, as the current clinical profile of Roche’s therapy is considered excellent.
Mim8 is projected to generate approximately USD 900 million in market revenue by 2036, highlighting its strong commercial potential, as per Javed. Javed further added that its approval could further expand the non-factor replacement therapy landscape by offering an additional prophylactic option with flexible dosing, advancing long-term disease management for patients with hemophilia A.
Late-Year Rare Disease Innovation: Regulatory Decisions and Pipeline Progress
The final months of 2026 are expected to bring another important regulatory milestone in rare diseases with Savara’s MOLBREEVI (molgramostim) for autoimmune pulmonary alveolar proteinosis (aPAP). The therapy is under FDA review as the potential first disease-specific treatment for aPAP, with the PDUFA date extended to November 22, 2026, following a major amendment to the BLA. Notably, the FDA has not identified any safety, efficacy, or manufacturing concerns during its review. Supported by Fast Track, Breakthrough Therapy, and Orphan Drug designations, Molbreevi has the potential to address a significant unmet clinical need.
Industry Expert Sentiments: Pulmonologists view the development of an FDA-approved version of inhaled GM-CSF as a positive step for the aPAP community, where few of them have high confidence that the next Phase III trial would be successful, while others are skeptical regarding changes in clinical trial measurements, noting it can be difficult to believe a drug will perform better simply by changing how results are measured after failing to reach targets initially
According to Aparna Thakur, Project Manager of Forecasting and Analytics at DelveInsight, MOLBREEVI is projected to generate approximately USD 1.1 billion in market revenue by 2036, underscoring its strong commercial potential in this underserved rare disease market.
BioMarin’s BMN 351 is advancing as a promising investigational therapy for Duchenne muscular dystrophy (DMD). Interim clinical data have demonstrated a favorable safety profile and encouraging dystrophin expression. Pending confirmatory data, BMN 351 is expected to compete with SGT-003 (Solid Biosciences; Phase III) and ifetroban (Cumberland Pharmaceuticals; Phase II) in the evolving DMD treatment landscape. In fact, BMN 351 is often compared to Dyne Therapeutics’ DYNE-251, while DYNE-251 offers less frequent monthly dosing (Q4W), early comparisons suggest BMN 351’s 5% expression at 9 mg/kg (weekly) is competitive with DYNE-251’s reported levels at similar timepoints. BioMarin estimates roughly 10,000 patients eligible for BMN 351 globally.
Industry Expert Sentiments: BMN 351 and other next-generation oligonucleotides (like those from Dyne and Entrada) could become stable, cash-generating assets as the market moves toward more effective disease-modifying treatments, however, physicians remain cautious about the limited clinical benefits of existing exon skippers
Commercial Outlook
The H2 2026 FDA calendar highlights the industry’s continued shift toward precision medicine, disease-modifying therapies, patient-centric innovations, and rare disease treatments. Several first- and best-in-class candidates are poised to reshape their respective markets while intensifying competition across multiple therapeutic areas.
Among the key commercial opportunities, TRUTAKNA could redefine the IgA nephropathy market, while gedatolisib has the potential to become the first targeted therapy for HR-positive, HER2-negative, PIK3CA wild-type advanced breast cancer. Subcutaneous KEYTRUDA is expected to strengthen Merck’s immuno-oncology franchise, whereas bictegravir plus lenacapavir could reinforce Gilead’s leadership in HIV through a simplified, high-barrier maintenance regimen.
Precision oncology remains a major growth driver, with zidesamtinib addressing unmet needs in ROS1-positive NSCLC and RP1 advancing oncolytic virotherapy in advanced melanoma. In diagnostics, MK-6240 could accelerate biomarker-guided management of Alzheimer’s disease.
Innovation in hematology and rare diseases is also expected to drive market expansion. Denecimig (Mim8) could broaden prophylactic options in hemophilia A, Molbreevi may become the first disease-specific therapy for aPAP, and BMN 351 highlights the continued advancement of genetic medicine for DMD
As per Thakur, several of these therapies are projected to achieve strong commercial success, including TRUTAKNA (USD 1.6 billion by 2036), MOLBREEVI (USD 1.1 billion by 2036), Mim8 (approximately USD 900 million by 2036), and zidesamtinib (approximately USD 400 million by 2036).
Collectively, the anticipated FDA decisions in H2 2026 are expected to accelerate innovation, intensify market competition, and expand treatment options, making the second half of 2026 a pivotal period for the global biopharmaceutical industry.
Downloads
Article in PDF
Recent Articles
- Ultomiris Receives US Priority Review for Adult IgA Nephropathy Treatment; FDA Approves WELIREG P...
- Ipsen to Buy Epizyme; BioMarin’s Gene Therapy for Hemophilia; AbbVie’s Qulipta for Ch...
- SNIPPET
- Daiichi Sankyo’s Ezharmia; Pfizer & Sangamo Hemophilia A Gene Therapy Trial; Approval for Fe...
- The Future is Here: BioMarin’s Roctavian First Hemophilia A Gene Therapy Paving the Way for a Cure



