An in-depth Assessment of the Top Drugs Launched by Leading Global Companies in the First Half of 2021 (H1)
Nov 01, 2021
Table of Contents
Progress is driven by innovation. When it comes to developing novel medications and therapeutic biological products, the FDA’s Center for Drug Evaluation and Research (CDER) assists the pharmaceutical sector at every stage of the process. CDER offers scientific and regulatory assistance needed to bring innovative medicines to market by understanding the research used to produce new products, testing and manufacturing techniques, and the illnesses and conditions that new products are meant to address. The novel FDA drug approval procedure enables researchers to establish the optimal dosage for both children and adults, find the optimum route of administration, and screen for potential drug interactions. An FDA approval indicates that the drug’s safety and efficacy resulted in benefits that outweigh known and potential concerns for the targeted population. Every new year ushers forth a fresh crop of medication approval candidates. However, the expectation is especially strong this year, given the fresh optimism for success following 2020’s disaster year. Aside from the industry’s remarkable response to COVID-19, pharma developers have advanced ground-breaking medicines for conditions impacting millions of people worldwide. On this note, let’s have a look at the most anticipated drug launches in the first half of 2021 (H1).
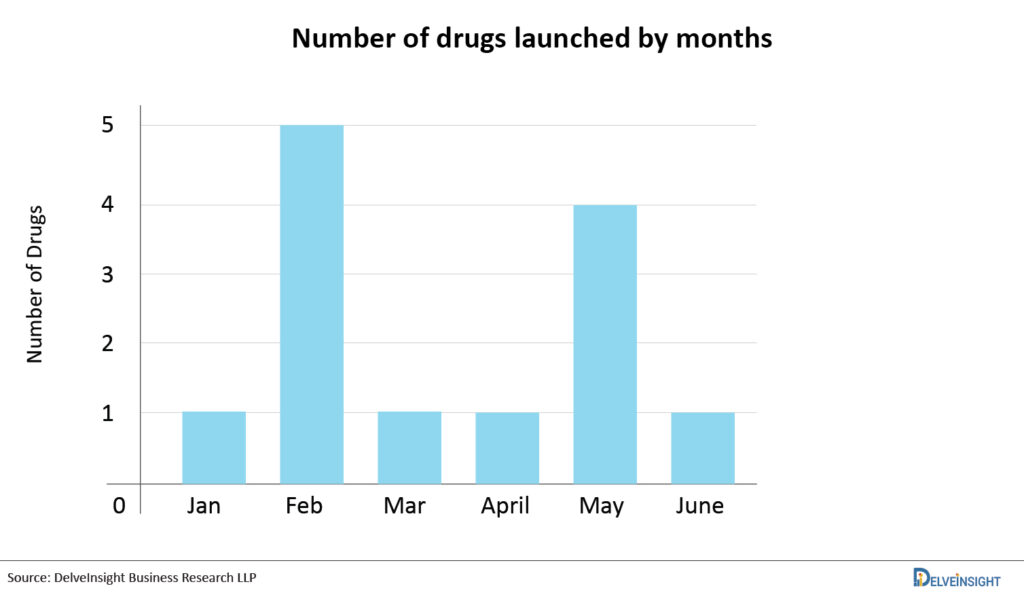
Lupykynis: Aurinia Pharmaceuticals: Lupus (January)
In January 2021, FDA approved Lupkynis (voclosporin), an oral drug for the treatment of adult patients with active lupus nephritis developed by Aurinia Pharmaceuticals. A novel, structurally modified calcineurin inhibitor, acts as an immunosuppressant that binds and inhibits calcineurin. By blocking calcineurin, a signaling protein involved in T cell activation, the small molecule oral drug is designed to inhibit IL-2 and tamps down inflammation in the kidney.
Downloads
Click Here To Get the Article in PDF
Recent Articles
- Teva sells generics; Valeant trades; Roche’s Genentech; J&J’s next potential fro...
- Notizia
- ASH 2022: Roche’s Polivy vs ADC Therapeutics’ Zynlonta, Antibody-Drug Conjugates (ADC) in R/R DLBCL
- FDA Approves LUMAKRAS with VECTIBIX for KRAS G12C-Mutated Colorectal Cancer; PYC Receives FDA Rar...
- Sarepta Therapeutics’s SRP-9001 Gene Therapy; FDA Approves Astellas’ VEOZAH; FDA Orphan Drug Desi...
In December 2019, Aurinia Pharmaceuticals announced positive efficacy and safety results from its pivotal AURORA Phase III trial of voclosporin, in combination with mycophenolate (MMF) and low-dose corticosteroids, for lupus nephritis treatment. Patients receiving LUPKYNIS had a 50% reduction in urine protein creatinine ratio (UPCR) twice as quickly as those who received SoC. A greater proportion of LUPKYNIS-treated patients had a complete renal response at 24 weeks compared with those who received SoC. Patients administered with LUPKYNIS had better response rates in all parameters in all immunologically active groups of Lupus Nephritis studied. The drug has the inherent advantage over legacy CNIs such as cyclosporine or tacrolimus due to its flat-dosing, thereby eliminating the need for therapeutic drug monitoring.
The drug had obtained fast track designation from the FDA in March 2016 and in June 2021, Aurinia Pharmaceuticals announced that the Company’s licensing partner, Otsuka Pharmaceutical Europe, filed an initial Marketing Authorization Application for voclosporin for the lupus nephritis treatment to the EMA. Lupkynis is also being evaluated in other indications such as Noninfectious Uveitis, Keratoconjunctivitis Sicca, Kidney Diseases, and Dry Eye Syndrome. Aurinia Pharma’s key competitors include Bristol Myers Squibb’s deucravacitinib, Roche-Genentech’s Gazyva, Janssen’s Tremfya, Novartis’ Cosentyx, and AstraZeneca’s anifrolumab.
“Lupkynis is the first and only oral treatment in the lupus nephritis market. The company has set a higher standard for those repurposing voclosporin for lupus nephritis. Lupkynis from Aurinia is expected to compete with Benlysta from GlaxoSmithKline. It is worth mentioning that Lupkynis has a slight advantage of oral administration, however its label has the following Black Box Warning of Malignancies and Serious Infections. Lupus patients will have a promising future with more options in coming years, as companies such as Roche (Gazyva; lupus nephritis) and AstraZeneca (Anifrolumab; systemic lupus erythematosus) also intend to enter the market.”
Tepmetko: Merck: Lung Cancer (February)
In February 2021, the USFDA approved Merck’s Tepmetko (Tepotinib) for the treatment of adult patients with metastatic NSCLC harboring mesenchymal-epithelial transition (MET) exon 14 skipping alterations. It is the first and only FDA-approved MET inhibitor that offers once-daily oral dosing and is administered as two 225 mg tablets (450 mg). The European Medicines Agency (EMA) has validated the application for tepotinib for the treatment of adult patients with advanced NSCLC (METex14 skipping) for review, as announced in November 2020.
It is a kinase inhibitor that targets MET, including variants with exon 14 skipping alterations, and inhibits hepatocyte growth factor (HGF)-dependent and -independent MET phosphorylation and MET-dependent downstream signaling pathways and also inhibited melatonin 2 and imidazoline 1 receptor at clinically achievable concentrations. In vitro, tepotinib inhibited tumor cell proliferation, anchorage-independent growth, and migration of MET-dependent tumor cells. In mice implanted with tumor cell lines with oncogenic activation of MET, including METex14 skipping alterations, tepotinib inhibited tumor growth, led to sustained inhibition of MET phosphorylation, and, in one model, decreased the formation of metastases.
This drug approval was based on results from the pivotal Phase II VISION study evaluating Tepmetko as monotherapy in patients with advanced NSCLC with METex14 skipping alterations The ORR was 43% among the treatment-naive patients, with a median DOR of 10.8 months. The ORR was also 43% among the previously treated patients, with a median DOR of 11.1 months. The FDA completed its review of TEPMETKO under its RealTime Oncology Review pilot program after previously granting Breakthrough Therapy Designation, orphan Drug Designation (ODD). In March 2020, TEPMETKO was initially approved in Japan for the treatment of advanced NSCLC patients with MET exon 14 skipping mutations, whether or not they’ve received prior therapy. This candidate is currently being investigated in Metastatic Colorectal Cancer (RAS/BRAF wt, MET amplified), NSCLC (EGFR mutant, MET amplified).
This is especially interesting given the active pipeline for c-met NSCLC products, including AstraZeneca/HUTCHMED (Savolitinib), Abion (ABN401), Apollomics (APL 101) Turning Point Therapeutics (TPX-0022), and understanding how well Tepmetko is addressing the unmet needs and where pipeline agents can expect to play will be critical as new products enter the space.
“In recent years, narrow biomarker-specific populations for NSCLC have become a hot topic, with several new treatments approved for biomarker-specific NSCLC. Pharmaceutical companies are also attempting to profit on the commercial opportunity by developing medications that target specific molecular aberrations. Two MET inhibitors, i.e.,Tabrecta and Tepmetko are such examples. Although Tepmetko received the first global clearance in Japan in 2020, Novartis beat Merck KGaA’s Tepmetko in the US market, as in February 2021 based on a phase II Vision Trial Tepmetko received a regulatory nod from FDA. Based on the phase II GEOMETRY mono-1 study, in 2020, Tabrecta (capmatinib) was approved as the first FDA-approved medication to treat NSCLC with specific mutations (those that cause mesenchymal-epithelial transition or MET exon 14 skipping). Merck is also trying to expand Tepmetko’s label by entering into different indications. While the market for MET mutation is not as vast as the market for EGFR and KRAS mutation, with the entry of approved and upcoming potential candidates, the market for MET mutation is going to boom in coming years.”
Rybrevant (amivantamab; Janssen) is the first fully-human, bispecific EGFR and c-Met antibody approved for the NSCLC treatment that targets EGFR exon 20 insertion mutations. It has the potential to benefit patients having Exon 20 mutation insertions, who are currently facing challenges with the currently available oral EGFR-targeted or immune checkpoint inhibitor therapies. It binds extracellularly (outside of the cell) inhibiting tumor growth and leading to tumor cell death. Janssen Research and Development, a Johnson & Johnson company (J&J), is responsible for the development of Rybrevant.
The US FDA has granted Breakthrough therapy designation to Rybrevant in 2020, and in December 2020 the US FDA also granted priority review to the BLA submitted for Rybrevant. In May 2021, on the basis of the BLA submitted, the US FDA granted accelerated approval to amivantamab-vmjw, a bispecific antibody directed against epidermal growth factor (EGF) and MET receptors, for adult patients with locally advanced or metastatic NSCLC with epidermal growth factor receptor (EGFR) exon 20 insertion mutations. The approval of Rybrevant was based on the CHRYSALIS trial (NCT02609776). Out of 81 patients, 40% of patients had an ORR towards the therapy and the average duration of the response was 11.1 months and in about 63% of patients, the response lasted for 6 months or longer. The company is also investigating the drug in combination with carboplatin-pemetrexed for patients with advanced or metastatic EGFR-mutated NSCLC with exon 20 insertion mutations (PAPILLON; NCT04538664; Phase III). Along with that, the drug is also being investigated in a comprehensive clinical development program for untreated advanced EGFR-mutated NSCLC treatment. The clinical trial evaluating the drug in combination with lazertinib in the treatment of patients with NSCLC characterized by EGFR exon 19 deletion or L858R mutations (CHRYSALIS-2; NCT04077463; Phase Ib), and recently in September 2021 the company presented preliminary results. Apart from these, there are several other trials investigating Rybrevant in the NSCLC treatment.
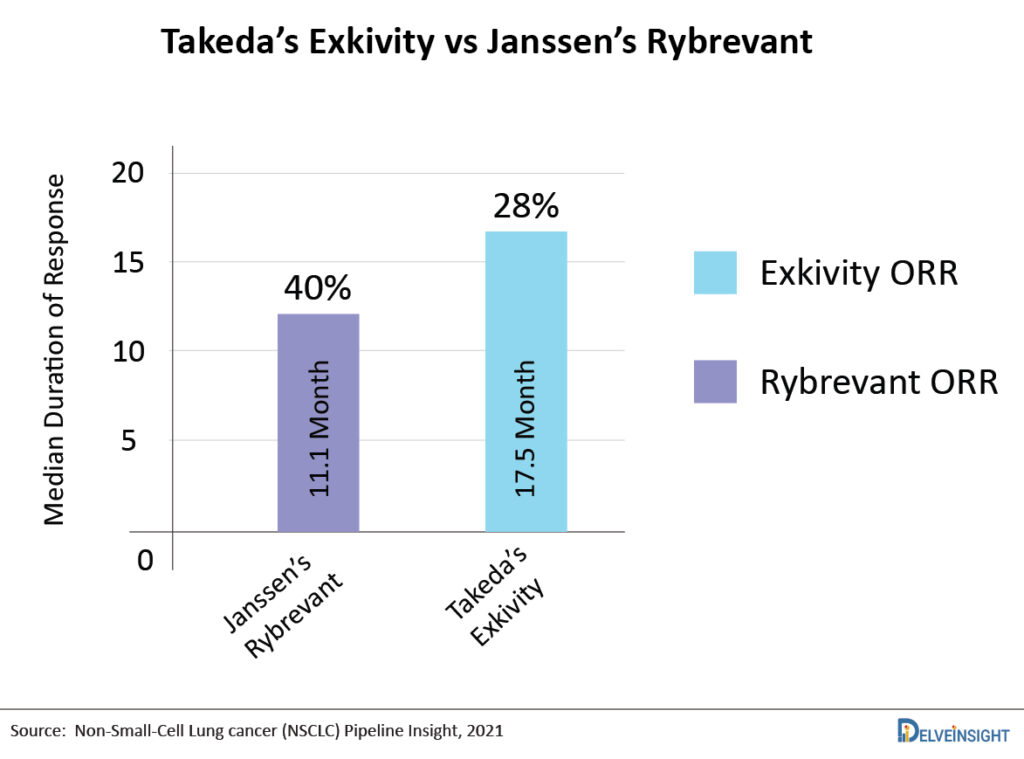
One of the major competitors of Rybrevant will be Takeda’s Exkivity as both candidates target the same subset of patients. Recent data presented by Takeda Exkivity showed 28% ORR in post-chemo EGFR exon 20 insertion patients by analysis of an independent data review committee, and Investigator-assessed ORR was 35%, whereas Rybrevant demonstrated an ORR of 40%. However, the patients on treatment with Exkivity experienced mDOR 17.5 months, whereas for patients with Rybrevant the mDOR was 11.1 months. Both the companies are also planning to enter in newly diagnosed NSCLC with EGFR exon 20 alterations setting.
Patients with EGFR exon 20 mutations have a poor response to other EGFR inhibitors, such as AstraZeneca’s Tagrisso, because these drugs are not specifically designed to treat this disease subtype. So far, only Exkivity and J&J’s Rybrevant have been authorized by the FDA to treat the exon 20 insertion mutation. Rybrevant had a monopoly over the EGFR exon 20 mutations target pool for a short time, however that changed with the entry of Takeda’s Exkivity. Janssen presented positive Phase I Results for RYBREVANT for the Treatment of Advanced NSCLC With MET Exon 14 Skipping Mutations at the International Association for the Study of Lung Cancer’s (IASLC) 2021 World Conference on Lung Cancer (WCLC), indicating that Rybrevant has broader efficacy against both EGFR and MET-driven tumors. The market for Rybrevant is projected to flourish as the company expands its label to other mutations and earlier lines of settings.
Lumakras (Sotorasib): Amgen: Lung Cancer (May)
Sotorasib is an inhibitor of KRASG12C, a tumor-restricted, mutant-oncogenic form of the RAS GTPase, KRAS. Sotorasib forms an irreversible, covalent bond with the unique cysteine of KRASG12C, locking the protein in an inactive state that prevents downstream signaling without affecting wild-type KRAS. It blocked KRAS signaling, inhibited cell growth, and promoted apoptosis only in KRAS G12C tumor cell lines.
In May 2021, the FDA granted expedited approval to Lumakras (sotorasib; Amgen), a RAS GTPase family inhibitor, for adult patients with KRAS G12C mutant locally advanced or metastatic NSCLC who have had at least one previous systemic treatment, as indicated by an FDA-approved test. Sotorasib was granted Breakthrough Therapy Designation in the US. KRAS is an oncogene that makes a protein involved in cell signaling pathways. The natural, unchanged form of the gene is called wild-type KRAS. Mutated forms of the KRAS gene have been found in some types of cancer, including non-small cell lung cancer, colorectal cancer, and pancreatic cancer.
Amgen had conducted one of the broadest global KRASG12C programs with eight regulatory submissions for advanced NSCLC evaluating Lumakras as monotherapy and in combination with docetaxel or an anti-PD-1/L1. The company presented its results from CodeBreak 100 trial during ASCO 2021, on the basis of which the drug was approved in second-line NSCLC. Interestingly, the study showed a durable response rate of 37.1%, a disease control rate of 80.6%, and a median progression-free survival of 6.8 months.
Amgen’s Lumakras is expected to compete with Mirati’s Adagrasib, which is being evaluated as both monotherapy and in combination with Cetuximab. Compared with Lumakras’ 37% response rate, adagrasib has triggered responses in 45% of patients in its Phase I/II study, although in a relatively smaller clinical trial. Amgen is also evaluating Lumakras in combination with other targets that inhibit MEK, SHP2, EGFR, mTOR, and CDK mutations.
Apart from NSCLC, Amgen and Mirati move head-to-head, targeting metastatic colorectal cancer (mCRC) as their second indication. However, as per recent news published, Amgen has effectively given up with Lumakras in colorectal cancer after reporting an ORR of just 7%. In NSCLC, both adagrasib and sotorasib seem evenly matched, showing 35–45% remission rates, making them a probable historic milestone in lung cancer therapy. However, we remain unsure about Lumakras’ success in colorectal cancer, which could be an opportunity for Mirati as the colorectal cancer cohort of its Krystal-1 study has shown an ORR of 17%, which provides an advantage for the company in the Colorectal KRAS niche.
In addition, there is a list of pharma companies queuing up to Amgen and Mirati, including Cardiff Oncology, Verastem, Onconova Therapeutics/Bristol-Myers Squibb, Boehringer Ingelheim, Revolution Medicines, Novartis Pharmaceuticals, InventisBio Inc./Merck Sharp & Dohme Corp and Moderna/Merck are working to advance their respective KRAS inhibitors already in clinical trials. Out of the list of Amgen and Mirati’s competitors, Cardiff Oncology looks the most promising one with its recently published positive Phase Ib/II dataset.
“Lumakras is the first KRAS G12C inhibitor to show an overall survival benefit, and the data represent a major step forward for patients with KRAS G12C-mutated NSCLC where the standard of care options are suboptimal. Before the approval of LUMAKRAS, the treatment of advanced NSCLC patients with the KRASG12C mutation was unfulfilled due to a lack of therapy options and poor results. Even though KRAS is the most frequently mutated gene in NSCLC, pharmaceutical firms have had little success with it, and it is deemed an undruggable target. However, in May 2020, the US FDA granted accelerated approval of sotorasib. Expanding its label to the earlier line of setting might result in a monetary boom for Amgen. In patients with KRAS p.G12C–mutated NSCLC, updated data from the CodeBreaK100 study demonstrated durable therapeutic benefit. Mirati presented new data at ESMO 2021 that suggested their investigational KRAS-candidate Adagrasib could outperform Amgen’s Lumakras, at least in colon cancer treatment, despite the fact that Lumakras is approved for lung cancer but not colon cancer. Mirati intends to deliver updated NSCLC data in the second half of 2021 (H2) and file an NDA for Adagrasib in the fourth quarter of 2021 in the United States for the possible treatment of patients with NSCLC who have the KRASG12C mutation and have received prior systemic therapy. After a positive regulatory nod, Adagrasib can give a stiff competition to Lumakras.”
Abecma (Idecabtagene vicleucel/bb2121/ ide-cel): Bristol-Myers Squibb/Bluebird bio: Multiple Myeloma (March)
Abecma (ide-cel) is a B-cell maturation antigen (BCMA)-directed genetically modified autologous chimeric antigen receptor (CAR) T cell immunotherapy co-developed, and co-promoted by Bristol Myers Squibb and Bluebird bio and is only approved for Multiple Myeloma treatment. It is the first and only approved CAR T cell therapy that is directed to recognize and bind to BCMA, leading to the death of BCMA-expressing cells.
Abecma first received its approval in March 2021, for the treatment of adult patients with multiple myeloma who have not responded to, or whose disease has returned after, at least four prior lines (different types) of therapy. In August 2021, European Commission granted Conditional Marketing Authorization, for relapsed and refractory multiple myeloma treatment in adults, who have received at least three prior therapies, including an immunomodulatory agent, a proteasome inhibitor, and an anti-CD38 antibody, and have demonstrated disease progression on the last therapy. This fourth-line indication contrasts with that in the US, where the therapy is approved for fifth-line use. However currently, Abecma is available only through a restricted program under a Risk Evaluation and Mitigation Strategy (REMS) called the ABECMA REMS in the US, this is because Abecma carries the risk of serious side effects that can be fatal or life-threatening and come with a black box warning. Although the approval of Abecma is indicated for patients who have had at least four prior lines of therapy (US), there are multiple studies currently underway evaluating it in the second-and third-line setting.

The approval of the therapy was granted on the basis of the Phase II KarMMa study (NCT03361748), which evaluated Abecma in 128 patients with heavily pre-treated and highly refractory multiple myeloma. In the Phase II KarMMa trial, the overall response rate (ORR) for the efficacy evaluable population was 72% in multiple myeloma patients receiving the CAR-T therapy (median DoR: 11 months), with 28% of patients achieving a stringent complete response (sCr), with a median DoR of 19 months. Of the patients who achieved sCR, an estimated 65% had remission lasting at least 12 months. Previously, the US FDA granted orphan drug designation (2016) and breakthrough therapy designation (2017) to the therapy and the EMA granted orphan (2017) as well as prime designation (2017).
“Despite receiving clearance months later than expected, Bristol Myers Squibb and Bluebird are still ahead of Johnson and Johnson’s and Legend’s Biotech, as they are also developing a competing CAR-T treatment for multiple myeloma known as ciltacabtagene autoleucel (cilta-cel). The company has filed the BLA application to the US FDA in April 2021 and has received a priority review, setting up an FDA decision by November 29, 2021. If approved, Cilta-cel would definitely give a tough competition to Abecma and would narrow down the lead of Abecma, thus dividing the market share of Abecma. Until the approval of Cilta-cel, Abecma can definitely enjoy its market presence as the first and only approved CAR T cell therapy for Multiple Myeloma.
“As per DelveInsight’s Multiple Myeloma market research, among the promising emerging therapies, Bristol Myers Squibb and Bluebird Bio’s anti-BCMA CAR T-cell therapy is expected to generate the maximum revenue owing to promising results. By 2030, Abecma is projected to generate sales of USD 1,558 million in the 8MM, i.e., US, EU-5, JP, and China.”
Unfortunately, the encouraging findings of anti-BCMA CAR T-cell treatments are not without their own set of difficulties. Aside from BCMA CAR-T products, BCMA bispecific antibodies are also being tested in clinical trials. Aside from cilta-cel CAR T-cell therapy, Regeneron’s REGN5458 and Janssen’s Teclistamab (formerly JNJ-64007957) bispecific antibodies potentially pose a threat to Abecma. Competitors like Poseida Therapeutics, CARsgen Therapeutics, Precision BioSciences, Cartesian Therapeutics, and others are also developing CAR-T therapies for Multiple Myeloma, however, they are in an early stage of development and would take time to enter the market.
Truseltiq : Bridge Bio: Cholangiocarcinoma (May)
Truseltiq is a kinase inhibitor indicated for the treatment of adults with previously treated, unresectable locally advanced, or metastatic cholangiocarcinoma with a fibroblast growth factor receptor 2 (FGFR2) fusion or other rearrangements. Cholangiocarcinoma affects roughly 20,000 people in the United States each year, with a five-year survival rate of only 9%.
In May 2021, the US FDA granted accelerated approval to Truseltiq (infigratinib) for the treatment of adults with previously treated, unresectable locally advanced, or metastatic cholangiocarcinoma with an FGFR2 fusion or other rearrangements. The FDA also approved FoundationOne CDx (Foundation Medicine) as a companion diagnostic test to select patients for treatment with infigratinib. The approval of the drug was based upon a Phase II study. In the multicenter, open-label, single-arm, Phase II study—called CBGJ398X2204 (NCT02150967)—108 patients who had undergone at least one treatment for advanced cholangiocarcinoma received 125 mg of infigratinib daily for 21 days of 28-day cycles until unacceptable toxicity or disease progression. The study’s primary endpoint demonstrated a confirmed ORR of 23%, with one complete response and 24 partial responses. The study also showed a median DOR of 5.0 months (3.7-9.3 months). Among the 23 responders, eight patients maintained the response for six months or more. The most common side effect of the drug was hyperphosphatemia, which affected around 77% of patients.
Previously in January 2020, the US FDA has granted Fast Track Designation for treatment in adults with first-line advanced or metastatic Cholangiocarcinoma and Orphan Drug Designation for cholangiocarcinoma treatment. At present, Truseltiq is not FDA approved for any other indication in the United States and is not approved for use by any other health authority. Apart from cholangiocarcinoma, Truseltiq is also being evaluated in other indications such as Urothelial Carcinoma, Achondroplasia, and Gastric Cancer, and other FGFR-Driven Tumors. QED Therapeutic’s key competitors include Taiho Oncology’s Futibatinib, Basilea Pharmaceutica’s Derazantinib, and Hutchison Medipharma’s HMPL-453.
“As per the current scenario, the available advanced or metastatic Cholangiocarcinoma therapeutic options is very limited. Although targeted therapies have changed the treatment landscape, and survival outcomes in various types of cancer, cholangiocarcinoma patients have faced a lack of therapy options for many years. With the entry of innovative drugs, the market size of cholangiocarcinoma may expand. Prior to approval of Truseltiq, Incyte’s Pemazyre (pemigatinib) became the first approved targeted therapy for cholangiocarcinoma in 2020. Thus Truseltiq, the second approved targeted therapy, will be a promising therapeutic option. It is worth highlighting that Pemazyre and Truseltiq are FGFR2 inhibitors; however, Truseltiq’s pricing is cheaper than Pemazyre’s, giving Truseltiq a cost advantage over Pemazyre, perhaps attracting a larger patient group.”
Evkeeza : Regeneron: Hypercholesterolemia (February)
Regeneron’s Evkeeza (evinacumab) is a fully recombinant human monoclonal antibody invented using the company’s VelocImmune technology. The same technology behind Dupixent, Libtayo, Praluent, Kevzara, and Inmazeb.
In February 2021, US FDA approved Evkeeza as an adjunct to other low-density lipoprotein cholesterol (LDL-C) lowering therapies to treat adult and pediatric patients aged 12 years and older with homozygous familial hypercholesterolemia (HoFH). Evkeeza is the first FDA-approved treatment that binds to and blocks the function of angiopoietin-like 3 (ANGPTL3), a protein that plays a key role in lipid metabolism. The FDA evaluated Evkeeza under Priority Review, following the decision in 2017 to grant Evkeeza Breakthrough Therapy designation and Orphan Drug designation a year prior to that.
The approval carries the limitations that the effectiveness and safety of Evkeeza have not been established in patients with other causes of hypercholesterolemia, including those with heterozygous familial hypercholesterolemia (HeFH), and that the effects of Evkeeza on cardiovascular morbidity and mortality have not been determined.
In June 2020, the EMA’s Committee for Medicinal Products for Human Use (CHMP) recommended an accelerated assessment for Evkeeza based on the high unmet medical need and therapeutic innovation demonstrated by the product. The European Medicines Agency, therefore, decided that Evkeeza’s benefits are greater than its risks, and it can be authorized for use in the EU. Therefore, Evkeeza has been authorized under ‘exceptional circumstances’.
Currently, Regeneron is evaluating Evkeeza in refractory hypercholesterolemia and severe hypertriglyceridemia. Evkeeza is expected to face competition from PCSK9 inhibitor and rival Amgen’s Repatha, which is already available in the HoFH market since 2015. Apart from Evkeeza, Regeneron’s Praluent also received approval as an adjunct to other LDL-C-lowering therapies in adult patients with HoFH to reduce LDL-C in April 2020. We expect both Evkeeza and Praluent to compete with Amgen’s Repatha, however, Evkeeza’s high cost of therapy questions its accessibility to all the rare disease patients. As per our analysis, Evkeeza is expected to reach a peak sales of approximately 100 million USD in 7MM (US, EU5, and Japan) in HOFH patients only.
Considering that Evkeeza helped reduce patients’ triglyceride levels by 57% at median after 12 weeks, compared with an overall increase of 1.8% for patients taking placebo in a Phase II study, the drug is now heading towards its further speedy development in hypertriglyceridemia patients, which are millions of patients in numbers, and if approved, it is expected to garner a good share in the Hypertriglyceridemia market.
Ukoniq: TG Therapeutics: Lymphoma (February)
UKONIQ (umbralisib) is the first and only oral inhibitor of phosphoinositide 3 kinases (PI3K) delta and casein kinase 1 (CK1) epsilon. PI3K-delta is known to play an important role in supporting cell proliferation and survival, cell differentiation, intercellular trafficking, and immunity and is expressed in both normal and malignant B-cells. CK1-epsilon is a regulator of oncoprotein translation and has been implicated in the pathogenesis of cancer cells, including lymphoid malignancies. Rhizen Pharma discovered umbralisib, which was later licensed to TG Therapeutics at an IND stage in 2012.
It is indicated for relapsed or refractory marginal zone lymphoma (MZL) treatment in adults who have received at least one prior anti-CD20-based regimen and for the treatment of adult patients with relapsed or refractory follicular lymphoma (FL) who have received at least three prior lines of systemic therapy. UKONIQ was granted Breakthrough Therapy Designation for Marginal Zone Lymphoma treatment and orphan drug designation for MZL and Follicular Lymphoma treatment.
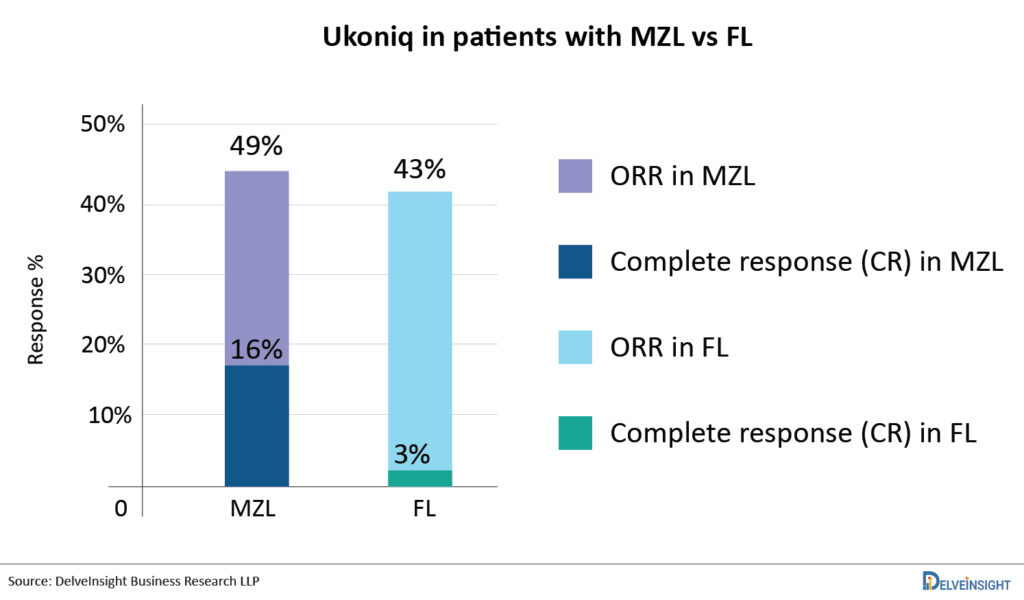
In February 2021, US FDA has approved Ukoniq for the treatment of adult patients with relapsed or refractory MZL who have received at least one prior anti-CD20 based regimen and adult patients with relapsed or refractory FL who have received at least three prior lines of systemic therapy. Ukoniq monotherapy was approved based on two single-arm cohorts from the Phase II UNITY-NHL clinical study (UTX-TGR-205/NCT02793583). The ORR for patients with MZL was 49%, with 16% achieving complete responses. The ORR for patients with FL was 43% (with 3% obtaining complete responses). The average DOR was 11.1 months. Umbralisib is now being tested in patients with NHL and CLL in combination with other agents such as ublituximab. In May 2021, TG Therapeutics announced the FDA acceptance of BLA for Ublituximab in combination with UKONIQ as a treatment for Patients with Chronic Lymphocytic Leukemia and Small Lymphocytic Lymphoma. The BLA submission was based on the results of the Phase III UNITY-CLL trial. The US FDA previously granted Fast Track status to the combination of ublituximab and UKONIQ for CLL treatment in adults, as well as orphan drug designation to ublituximab in combination with UKONIQ for chronic lymphocytic leukemia treatment.
The company reported total net UKONIQ revenue generated from launch until the end of the second quarter of 2021 was USD 2.3 million. This medicine also received more than 90% of Medicare and commercial payers in the United States, and it was included in the NCCN Clinical Practice Guidelines for MZL and FL. With its clearance as the first oral, once-daily inhibitor, the PI3K class has been extended, and it may be a safer alternative than current players such as Gilead Sciences’ Zydelig, Bayer’ Aliqopa, and Secura Bio’s Copiktra. Not only that, but the drug’s long-term advantages, along with a favourable safety profile, offer up the potential of combinations. The company is already attempting to seek FDA approval in combination with ublituximab for CLL. The competitive landscape of PI3K Targeted Therapies is robust with companies like Incyte Corporation (Parsaclisib), MEI Pharma (Zandelisib), Rhizen Pharmaceuticals (Tenalisib/RP-6530), and others are investigating their drug candidates for NHL and other hematological malignancies. Other drugs classes approved or under development for the same subset can be a challenge for UKONIQ.
Zynlonta: ADC Therapeutics: Lymphoma ( April)
Zynlonta (loncastuximab tesirine), developed by ADC Therapeutics is a CD19-directed antibody and alkylating agent conjugate indicated for the treatment of adult patients with relapsed or refractory large B-cell lymphoma after two or more lines of systemic therapy, including diffuse large B-cell lymphoma (DLBCL) not otherwise specified, DLBCL arising from low-grade lymphoma, and high-grade B-cell lymphoma. Zynlonta is the first and only CD19-targeted antibody-drug conjugate (ADC) for relapsed/refractory DLBCL after two or more lines of systemic therapy. In June 2017, the therapy was granted orphan drug designation by the US FDA, and recently in July 2021, the EU has adopted a positive opinion for granting an orphan drug designation to Loncastuximab Tesirine.
In April 2021, the US FDA has granted accelerated approval to Zynlonta for the treatment of adult patients with relapsed/refractory large B-cell lymphoma following 2 or more lines of systemic therapy, including diffuse large B-cell lymphoma (DLBCL) not otherwise specified, DLBCL arising from low-grade lymphoma, and high-grade B-cell lymphoma. Just two weeks after getting accelerated FDA clearance, Zynlonta was added to the National Comprehensive Cancer Network (NCCN) Guidelines with a Category 2A recommendation.
The FDA granted authorization based on findings from the LOTIS-2 Phase II Study. The trial’s outcomes showed an ORR of 48.3%, with a complete response (CR) rate of 24.1%, and a partial response (PR) rate of 24.1%, with a mDoR of 10.3 months. The most frequent adverse events (≥20%) in a pooled safety population were thrombocytopenia, elevated gamma-glutamyltransferase, neutropenia, anemia, hyperglycemia, transaminase elevation, fatigue, hypoalbuminemia, rash, edema, nausea, and musculoskeletal discomfort. In September 2021, Overland ADCT BioPharma (a joint venture with ADC Therapeutics) reported that the first patient in a key Phase II clinical trial in patients with relapsed or R/R DLBCL in China has been dosed with Zynlonta. According to the company, pivotal clinical trials include the Phase I/II LOTIS-3 clinical trial of Zynlonta in combination with ibrutinib for the treatment of relapsed or refractory DLBCL and Mantle cell lymphoma (MCL), and the Phase II clinical trial of Zynlonta for relapsed or refractory Follicular Lymphoma treatment. In addition, the company is undertaking a LOTIS-5 Phase III confirmatory trial with rituximab in second-line non-transplant eligible DLBCL patients.
This candidate is already getting a lot of attention. Following rapid US FDA clearance in April 2021, the company reported net sales of USD 3.8 million for the two months. Given that there is already competition from other CD19-targeted medicines such as the CAR-T therapies from Novartis (Kymriah), Gilead Sciences (Yescarta), and Bristol Myers Squibb (Breyanzi), ZYNLONTA’s launch performance was driven by its differentiated profile in treating an area of significant unmet medical need. Zynlonta addresses the current unmet need of the lack of treatment options for patients with R/R DLBCL, including those who have been heavily pretreated and have a difficult-to-treat disease. Patients with R/R DLBCL, including those who have been heavily pretreated and are difficult-to-treat disease, such as patients who do not respond to first-line or any prior line of therapy, or who have failed CAR-T therapy or stem cell transplant-Have high-grade B cell lymphoma, including double hit/triple hit genetics, will benefit from Zynlonta. The company also hopes to expand Zynlonta in earlier lines of setting, which would likely increase its market share and size in the future. Investors are undoubtedly betting on ADC’s long-term prospects in DLBCL. Roche’s Polivy (CD79b-directed ADC) has already been approved for third-line DLBCL, and the company plans to expand into earlier stages of the disease. With the approval of ADCs in DLBCL, it is projected that interest in research and development in this field would grow even more.
Breyanzi (Lisocabtagene maraleucel / liso-cel/JCAR017): Bristol-Myers Squibb: Lymphoma (February)
Breyanzi (lisocabtagene maraleucel), developed by Bristol-Myers Squibb, is a CD19-directed genetically modified autologous T cell immunotherapy, administered intravenously in defined composition at a precise dose of CD8 and CD4 CAR T cells which induced durable responses in poor-prognosis patients with R/R aggressive NHL after at least two prior therapies. It is only approved for DLBCL treatment.
The US FDA has granted Breakthrough Therapy designation, Orphan drug designation, and Regenerative Medicine Advanced Therapy (RMAT) Designation to Breyanzi, whereas in December 2016, the EMA awarded PRIME status to Breyanzi
In February 2021, the FDA approved Breyanzi to treat adult patients with certain types of DLBCL who have not responded to, or who have relapsed after at least two other types of systemic treatment. The approval of Breyanzi was based upon the data from the TRANSCEND NHL 001 trial, in which 268 patients with R/R DLBCL received Breyanzi. Among 192 patients in the main efficacy population, the ORR was 73%, of which 53% of patients had a complete response (CR). Breyanzi showed sustained responses in patients who attained a CR but did not reach the median duration of response. In addition, Grade ≥3 cytokine release syndrome and Grade ≥3 neurologic toxicities occurred in 4% and 12% of patients, respectively, after Breyanzi treatment. In March 2021, Japan’s Ministry of Health, Labour, and Welfare (MHLW) announced the approval of Breyanzi to treat patients suffering from R/R DLBCL and R/R follicular lymphoma. Breyanzi is currently filed in the EU.
It is also being investigated for DLBCL treatment transplant eligible (2nd line), non-transplant eligible (2nd line), Chronic Lymphocytic Leukemia (3rd line+), Follicular Lymphoma / Marginal Zone Lymphoma (3rd line+), Pediatric B-Cell Acute Lymphoblastic Leukemia (2nd line+), Primary CNS Lymphoma (2nd line+), High-Grade B-cell Lymphoma (1st line), Mantle Cell Lymphoma (3rd line+). In June 2021, the company announced positive topline results from TRANSFORM, a global, randomized, multicenter Phase III study evaluating Breyanzi as a second-line treatment in adults with DLBCL compared to salvage therapy followed by high-dose chemotherapy and HSCT, which is currently considered a gold standard treatment for these patients.
With this approval, Breyanzi became the third CAR-T cell therapy approved for DLBCL along with rival CAR-T therapies from Novartis (Kymriah) and Gilead Sciences (Yescarta) to treat DLBCL patients after two or more lines of systemic therapy. If Breyanzi is compared with Kymriah and Yescarta, all the three therapies come along with a black box warning, however, the side effect profile of Breyanzi appears to be less severe than Yescarta and Kymriah, which could enable outpatient treatment. On the other hand, the Breyanzi launched with a list price of USD 410,300 for the one-time treatment compared with USD 373,000 for Yescarta and Kymriah, this can be attributed to the higher manufacturing cost of Breyanzi.
“Breyanzi sales in the United States totaled around USD 17 million, as reported in the second quarter of 2021. Kymriah, Yescarta, and Breyanzi will undoubtedly compete for market share, as these medications are approved for the same setting, and all three are looking to expand their markets by pursuing earlier lines of treatment and additional indications. Yescarta and Breyanzi recently showed positive outcomes in the second-line setting, however, Novartis’ Kymriah reported negative results in the Phase III BELINDA study. Breyanzi will likely face competition from more than simply licensed CAR-T cell therapies. Several companies, including Genmab (Epcoritamab), Miltenyi Biomedicine (Zamtocabtagene autoleucel), Allogene Therapeutics (ALLO-501A and ALLO-647), and others are evaluating their treatments for DLBCL and will eventually enter the DLBCL market space in the coming years. There are new antibodies and ADCs for DLBCL, as well. Zynlonta (ADC Therapeutics) was approved by the FDA in April 2021, and it targets CD19. Monjuvi (MorphoSys/Incyte), a CD19-targeting drug, has also been approved for the treatment of DLBCL in combination with Revlimid. Other contenders are Polivy (anti-CD79b ADC; Genentech) and Xpovio (XPO1 inhibitor; Karyopharm Therapeutics).”
Empaveli: Apellis Pharmaceuticals: Hemoglobinuria (May)
Pegcetacoplan (APL-2) (Apellis Pharmaceuticals) is an investigational, targeted C3 inhibitor designed to regulate excessive complement activation, leading to the onset and progression of many serious diseases. Pegcetacoplan is a synthetic cyclic peptide conjugated to a polyethylene glycol polymer that binds specifically to C3 and C3b, thereby regulating the cleavage of C3 and the generation of downstream effectors of complement activation. In PNH, extravascular hemolysis (EVH) is facilitated by C3b opsonization while intravascular hemolysis (IVH) is mediated by the downstream membrane attack complex (MAC). Pegcetacoplan acts proximally in the complement cascade controlling both C3b-mediated EVH and terminal complement-mediated IVH. The drug was approved for paroxysmal nocturnal hemoglobinuria (PNH) in May 2021 and is currently being evaluated in other related indications such as Complement c3 Glomerulopathy (C3G), Membranoproliferative Glomerulonephritis, Geographic Atrophy Secondary to Age-Related Macular Degeneration, Neovascular Age-Related Macular Degeneration, and Amyotrophic Lateral Sclerosis.
Considering the result outcomes in PNH, the patients treated with EMPAVELI showed an average increase of 2.4g/dL in their hemoglobin levels, while patients treated with Soliris showed an average decrease of 1.5g/dL in their hemoglobin levels. The normalization rates for hemolysis were also higher in EMPAVELI-treated patients. In addition, around 85% of EMPAVELI-treated patients were transfusion-free, compared to 15% patients treated with Soliris.
Pegcetacoplan has been granted Fast Track designation for PNH treatment. Recently, Apellis submitted Marketing applications for pegcetacoplan to treat PNH to the FDA and the EMA. Additionally, the FDA granted the application Priority Review designation and set a target action date of May 2021. An opinion from the Committee for Medicinal Products for Human Use (CHMP) is expected in 2021. Apellis and Sobi entered a collaboration to develop and commercialize systemic pegcetacoplan in October 2020.
We expect Empaveli to compete with the already approved Alexion Pharma’s Soliris and Ultomiris in the PNH market. Moreover, it is the only C3 inhibitor being evaluated in a Phase II trial for C3G with an estimated market size of more than USD 450 million in the 7MM by the year 2030. In C3G, Pegcetacoplan is expected to compete with Novartis’ Iptacopan, which is an oral capsule being evaluated in a Phase III trial for C3G.
Lybalvi: Alkermes: Schizophrenia (June)
Lybalvi, a combination of olanzapine and samidorphan, is approved for the treatment of adults with schizophrenia and/or bipolar I disorder. It is a once-daily, atypical antipsychotic that works as maintenance monotherapy for the acute treatment of manic or mixed episodes, or as an adjunct to lithium or valproate. It is an oral atypical antipsychotic combination of an established antipsychotic agent (olanzapine) and a novel μ-opioid receptor antagonist (samidorphan). Alkermes is responsible for the development of Lybalvi.
In June 2021, the US FDA approved Lybalvi for schizophrenia and bipolar 1 disorder treatment in adults. The approval of Lybalvi is based on results from 27 trials, including 18 studies assessing the drug and nine studies analyzing samidorphan alone. In the Enlighten clinical development program, Lybalvi demonstrated antipsychotic efficacy, safety, and tolerability, including statistically significantly less weight gain than olanzapine in patients with schizophrenia, which was reported from the ENLIGHTEN-2 study (NCT02694328). The approval of Lybalvi for the treatment of bipolar I disorder was supported by the established efficacy and safety profile of orally administered olanzapine in adequate and well-controlled studies. However, the label of Lybalvi comes along with a set of boxed warnings, regarding an increased risk of mortality in elderly patients with dementia-related psychosis. Thus, it is not approved for the treatment of patients with dementia-related psychosis. Along with that samidorphan, can cause withdrawal syndrome in patients physically dependent on opioids or lead to opioid overdose among those who try to overcome the opioid-blocking effect of samidorphan.

In addition, there is a list of pharma companies queuing up to develop a treatment for schizophrenia and/or bipolar I disorder. These companies include Otsuka Pharmaceutical (Brexpiprazole), Sunovion Pharmaceuticals (Ulotaront), Minerva Neurosciences (Roluperidone), Acadia Pharmaceuticals (Pimavanserin), Boehringer Ingelheim (BI 425809), and Avanir Pharmaceuticals (Deudextromethorphan).
Lybalvi is a promising new therapeutic option for persons suffering from schizophrenia or bipolar I disorder as it demonstrated a better safety profile than olanzapine. It also demonstrated less weight gain than traditional olanzapine. It is worth mentioning that it was not an easy journey to have this medicine approved by the FDA. Lybalvi was approved in the United States after an initial rejection by the US FDA due to manufacturing problems. The FDA’s approval is not the last roadblock in the way of the drug’s success. However, one of the major challenges which this drug will face is “lower-cost generics” that are available in the market.
Amondys 45: Sarepta: Duchenne Muscular Dystrophy (February)
Amondys 45 (casimersen) is the third exon skipping therapy approved by Sarepta for DMD in people who have a confirmed mutation of the dystrophin gene that is amenable to exon 45 skipping.
Amondys 45, which got accelerated approval in February 2021 and it is an antisense oligonucleotide and is based on the company’s proprietary phosphorodiamidate morpholino oligomer (PMO) based technology. The US FDA granted Fast Track, Priority Review designations and in April 2019 Orphan Drug Designation to this drug. Amondys 45 is approved under accelerated review based on increased dystrophin production in skeletal muscle of patients amenable to exon 45 skipping.
The commercialization of the drug began immediately and has roped in USD 7.1 million till June 2021. With the target patient pool of 8% of total DMD patients, and an approximate annual cost of therapy of USD 748,000 for patients weighing 30Kg, the drug is estimated to cross USD 400 million in revenue by 2030. As per Delveinsight’s analysis, Amondys 45 has currently no competition and is expected to face competition in case Daiichi’s DS-5141b, another exon 45 skipping therapy, gets approval in the United States. However, as per current findings, Daiichi is focusing on a first launch in Japan with a Phase II trial currently ongoing. With Daiichi in Japan and Sarepta in the US (potentially Europe in the future) a head-to-head competition is not expected so soon.
To expand the products market, the company is conducting a Phase III trial (ESSENCE) in Europe and the US. The same trial will be used for confirmation in the US for both casimersen and golodirsen (Vyondys 53). Moreover, the expansion of the drug in European markets will give a boost to product sales. However, in the past, no DMD therapy from Sarepta has been able to get EMA approval. Hopefully, Amondys 45 along with Vyondys 45 could break this trend.
What’s Next?
The availability of novel medications and biological products frequently means new treatment choices for patients and breakthroughs in health care. As a result, CDER encourages creativity and plays a critical role in advancing novel medication development. Every year, the CDER authorizes a wide range of novel pharmaceuticals and biological products. The US FDA approved many orphan drug designations, breakthrough therapy designations, and biologics licensing applications in the first half of this year H1. From big pharma companies to small, each launched different candidates for different conditions but almost half of the new drugs are for cancer and associated disease, with many benefiting from priority review and rapid approval processes. Amgen’s treatment for non-small cell lung cancer and Bristol Myers Squibb’s new CART-T therapy for multiple myeloma, as well as a cell therapy for large B-cell lymphoma, are among them. The first half of the year 2021 has been promising for the oncological sector as many companies investigated its candidate for various types of cancer. But we may witness some changes in the second half of the year as many pharma and biotech companies are evaluating their candidates for various dermatological, neurological, and immunological disorders. The second half will bring more novel treatment options for different conditions to improve patients’ lifestyles.
The top drugs that launched in the first half of 2021 (H1) include Lupykynis, Tepmetko, Rybrevant, Lumakras, Abecma, Truseltiq, Evkeeza, Ukoniq, Zynlonta, Breyanzi, Empaveli, Lybalvi, Amondys 45, and several others.
The leading companies that launched their lead assets in the first half of 2021 include Aurinia Pharmaceuticals, Merck, Janssen, Amgen, Bristol-Myers Squibb, Bluebird bio, Bridge Bio, Regeneron, TG Therapeutics, ADC Therapeutics, Apellis Pharmaceuticals, Alkermes, Sarepta, and others.
The leading players working diligently towards the development of novel CAR T-Cell Therapies for Multiple Myeloma include Bristol Myers Squibb, Bluebird Bio, Regeneron, Janssen, Poseida Therapeutics, CARsgen Therapeutics, Precision BioSciences, Cartesian Therapeutics, and others.
There is a list of pharma companies queuing up to develop a treatment for schizophrenia and/or bipolar I disorder. These companies include Otsuka Pharmaceutical (Brexpiprazole), Sunovion Pharmaceuticals (Ulotaront), Minerva Neurosciences (Roluperidone), Acadia Pharmaceuticals (Pimavanserin), Boehringer Ingelheim (BI 425809), and Avanir Pharmaceuticals (Deudextromethorphan).
There is a list of pharma companies queuing up to Amgen and Mirati, including Cardiff Oncology, Verastem, Onconova Therapeutics/Bristol-Myers Squibb, Boehringer Ingelheim, Revolution Medicines, Novartis Pharmaceuticals, InventisBio Inc./Merck Sharp & Dohme Corp and Moderna/Merck are working to advance their respective KRAS inhibitors already in clinical trials.
Downloads
Article in PDF
Recent Articles
- Lassen’s anti-IL-11 antibody; PTC’s COVID-19 trial; Lenzilumab’s result; Orca raises $192M
- Sanofi’s Rare Disease Drug Xenpozyme’ Approval; FDA Approves Novartis’ Pluvicto; AstraZenec...
- Daiichi Sankyo’s Trastuzumab Deruxtecan; ODD to Bexmarilimab for AML; Roche’ Alecensa; Ergomed Ai...
- Precigen’s PRGN-3006; Yescarta Approved as a First CAR T-cell Therapy for R/R LBCL; Biogen ...
- FDA Approves; AZ nabs; Nordisk settles; Novartis pays



